4.1 Anti-Arrythmic Drugs
Robi Islam
Introduction
Learning Outcomes
Normal cardiac conduction and electrophysiology
The heart functions via mechanical and electrical activity.
Mechanical activity refers to atrial and ventricular contraction, the mechanism by which blood is delivered to tissues. When deoxygenated blood returns to the heart via venous circulation, the blood enters the right atrium. Right atrial contraction and right ventricular pressure changes result in delivery of blood to the right ventricle through the tricuspid valve. Right ventricular contraction pumps blood through the pulmonic valve and through the pulmonary arteries to the lungs, where blood becomes oxygenated. The oxygenated blood then flows through the pulmonary veins into the left atrium. Left atrial contraction and left ventricle (LV) pressure changes result in delivery of blood through the mitral valve into the LV, contraction of which results in pumping of blood through the aortic valve and to the tissues of the body.
Mechanical activity is stimulated by the electrical activity of the heart. The heart possesses an intrinsic electrical conduction system.
 |
Normal myocardial contraction cannot occur without normal function of the heart’s electrical conduction system. Depolarization of the atria results in atrial contraction, and ventricular depolarization produces ventricular contraction. Perturbation of the heart’s electrical conduction system may result in dysfunctional atrial and/or ventricular contraction and may reduce cardiac output.
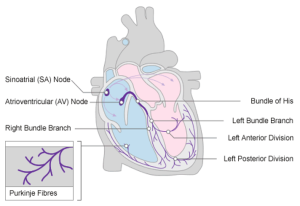
The process of normal cardiac conduction is summarised below:
- Action potentials generated by the sinoatrial (SA) node spread across the atria by cell-to-cell conduction, which leads to atrial contraction (i.e., the P wave)
- The atrioventricular (AV) node slows the electrical impulses considerably to allow enough time for complete atrial depolarisation and contraction prior to ventricular depolarisation
- The slowed electrical impulses enter the ventricle at the Bundle of His and then follow the left bundle branch (LBB) and right bundle branch (RBB)
- The LBB and RBB divide into an extensive network of Purkinje fibres that carry the impulses across the ventricles
- The result is rapid depolarisation of ventricular myocytes, that then leads to ventricular contraction.
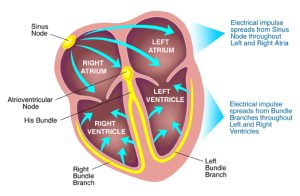 |
Cardiac Rate and Rhythm
As mentioned above, the chambers of the heart normally contract in a coordinated manner, pumping blood efficiently by a route determined by the valves. Coordination of contraction is achieved by a specialised conducting system. Normal sinus rhythm is generated by pacemaker impulses that arise in the sinoatrial (SA) node and are conducted in sequence through the atria, the atrioventricular (AV) node, bundle of His, Purkinje fibres and ventricles.
Cardiac cells owe their electrical excitability to voltage-sensitive plasma membrane channels selective for various ions, including sodium (Na+), potassium (K+) and calcium ions (Ca2+), the structure and function. The electrophysiological features of cardiac muscle that distinguish it from other excitable tissues include:
- pacemaker activity
- absence of fast Na+ current in SA and AV nodes, where slow inward Ca2+ current initiates action potentials
- long action potential (‘plateau’) and refractory period
- influx of Ca2+ during the plateau
Several of these special features of cardiac rhythm relate to Ca2+ currents. The heart contains intracellular calcium channels (i.e. ryanodine receptors and inositol trisphosphate-activated calcium channels, which are important in myocardial contraction) and voltage-dependent calcium channels in the plasma membrane, which are important in controlling cardiac rate and rhythm. The main type of voltage-dependent calcium channel in adult working myocardium is the L-type channel, which is also important in vascular smooth muscle; L-type channels are important in specialised conducting regions as well as in working myocardium.
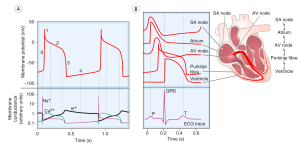
Action Potential
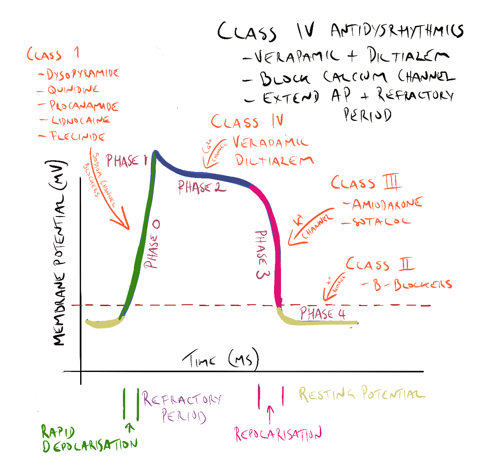
The action potential of an idealised cardiac muscle cell is divided into five phases: 0 (fast depolarisation), 1 (partial repolarisation), 2 (plateau), 3 (repolarisation) and 4 (pacemaker). Ionic mechanisms underlying these phases can be summarised as follows.
- Phase 0: rapid depolarisation, occurs when the membrane potential reaches a critical firing threshold (about −60 mV), at which the inward current of Na+ flowing through the voltage-dependent sodium channels becomes large enough to produce a regenerative (‘all-or-nothing’) depolarisation. Activation of sodium channels by membrane depolarisation is transient, and if the membrane remains depolarised for more than a few milliseconds, they close again (inactivation). They are therefore closed during the plateau of the action potential and remain unavailable for the initiation of another action potential until the membrane repolarises.
- Phase 1: partial repolarisation, occurs as the Na+ current is inactivated.
 |
- Phase 2: the plateau, results from an inward Ca2+ current. Calcium channels show a pattern of voltage-sensitive activation and inactivation qualitatively similar to sodium channels but with a much slower time course. The plateau is assisted by a special property of the cardiac muscle membrane known as inward-going rectification, which means that the K+ conductance falls to a low level when the membrane is depolarised. Because of this, there is little tendency for outward K+ current to restore the resting membrane potential during the plateau, so a relatively small inward Ca2+ current suffices to maintain the plateau. A persistent sodium current (INap) also contributes to the plateau; it is very small compared with the fast component of sodium current, but as it flows throughout the action potential it makes a substantial contribution to sodium loading during each cardiac cycle and is a major contributor to ischaemic arrhythmias and a drug target.
- Phase 3: repolarisation, occurs as the Ca2+ current inactivates and a delayed outwardly rectifying K+ current activates, causing outward K+ current. This is augmented by another K+ current, which is activated by high intracellular Ca2+ concentrations during the plateau.
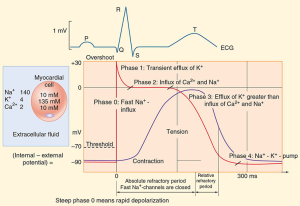 |
- Phase 4: the pacemaker potential, is a gradual depolarisation during diastole. Pacemaker activity is normally found only in nodal and conducting tissue. The pacemaker potential is caused by a combination of increasing inward currents and declining outward currents during diastole. It is usually most rapid in cells of the SA node, which therefore acts as a pacemaker for the whole heart. Cells in the SA node have a greater background Na+-conductance than do atrial or ventricular myocytes, leading to a greater background inward current. In addition, inactivation of voltage-dependent calcium channels wears off during diastole, resulting in increasing inward Ca2+ current during late diastole. Activation of T-type calcium channels during late diastole contributes to pacemaker activity in the SA node. The negative membrane potential early in diastole activates a cation channel that is permeable to Na+ and K+, giving rise to another inward current.
Refractory Period
After an impulse is initiated and conducted, there is a period during which cells and fibers cannot be depolarized again, which is referred to as the absolute refractory period (see Figure) and corresponds to phases 1, 2, and approximately one-third of phase 3 repolarization. The absolute refractory period also corresponds to the period from the Q wave to approximately the first half of the T wave on the ECG.
- During this period, if there is a premature stimulus for an electrical impulse, this impulse cannot be conducted because the tissue is absolutely refractory to conduction. However, there is a period following the absolute refractory period during which a premature electrical stimulus can be conducted and is often conducted abnormally, which is called the relative refractory period, corresponding roughly to the latter two-thirds of phase 3 repolarization on the action potential and to the latter half of the T wave on the ECG. The relative refractory period is also sometimes referred to as the “vulnerable period.” If a premature electrical stimulus is initiated during the relative refractory period, it can be conducted abnormally, potentially resulting in an arrhythmia.
📺 Watch the brief vodcast on the review of action potential and cardiac rhythm (14 minutes)
Arrhythmias – Disturbances of Cardiac Rhythm
Clinically, dysrhythmias are classified according to:
- the site of origin of the abnormality – atrial, junctional or ventricular;
- whether the rate is increased (>100 beats per minute [bpm] – tachycardia) or decreased (<60 bpm – bradycardia).
Cardiac arrhythmias are caused by (a) abnormal impulse initiation, (b) abnormal impulse conduction, or (c) both.
Abnormal initiation
Abnormal initiation of electrical impulses occurs due to abnormal automaticity. A decrease in sinus node automaticity results in a reduced rate of impulse generation and a slow heart rate (sinus bradycardia). Conversely, an increase in sinus node automaticity results in an increased rate of impulse generation and a rapid heart rate (sinus tachycardia). If other cardiac fibers become abnormally automatic, such that the rate of spontaneous impulse initiation exceeds that of the sinus node, or premature impulses are generated, other tachyarrhythmias may occur. Early after depolarisation (EAD) occurs during phase 2 (Ca2+ channels) or phase 3 (partially recovered Na+ channels) whereas delayed afterdepolarisation (DAD) occurs during phase 4 (high intracellular Ca2+ and inward Na+ current). Many cardiac fibers possess the capability for automaticity, including atrial tissue, the AV node, the Purkinje fibers, and ventricular muscle. In addition, fibers with the capability of initiating and conducting electrical impulses are present in left atrial myocardial sleeves that extend into the pulmonary veins. Abnormal atrial automaticity may result in premature atrial depolarizations or may precipitate atrial tachycardia or atrial fibrillation (AF); abnormal AV nodal automaticity may result in “junctional tachycardia” (so named because the AV node is also sometimes referred to as the AV junction). Abnormal automaticity originating from the pulmonary veins is a precipitant of AF. In addition, abnormal automaticity in the ventricles may result in premature ventricular complexes (PVCs) or may precipitate ventricular tachycardia (VT) or ventricular fibrillation (VF).
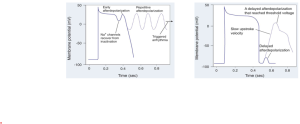 |
The mechanism of abnormal impulse conduction is referred to as reentry. Reentry is often triggered as a result of an abnormal premature electrical impulse (abnormal automaticity); therefore, in these situations, the mechanism of the arrhythmia is both abnormal impulse formation (automaticity) and abnormal impulse conduction (reentry). For reentry to occur, three conditions must be present. There must be: (a) at least two pathways down which an electrical impulse may travel (which is the case in most cardiac fibers); (b) a “unidirectional block” in one of the conduction pathways (this “unidirectional block” reflects prolonged refractoriness in this pathway, or increased “dispersion of refractoriness,” defined as substantial variation in refractory periods between cardiac fibers); and (c) slowing of the velocity of impulse conduction down the other conduction pathway.
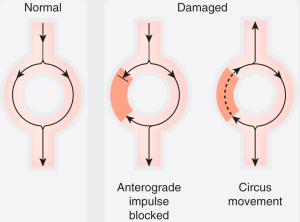 |
📺 Watch the brief vodcast on Arrhythmias (9 minutes)
Exercises: Check your understanding: Cardiac cycle, cardiac muscle and electrical activity, cardiac physiology
Antiarrhythmics
The antiarrhythmic drugs are used to treat and prevent cardiac arrhythmias which can be categorized into two main groups:
- Bradyarrhythmia including atrial-ventricular (AV) block, sick sinus syndrome and sinus bradycardia caused by drugs, hypothyroidism and myocardial function
- Tachyarrhythmia such as sinus tachycardia, atrial fibrillation (AF), atrial flutter, supraventricular tachycardia (SVT), reentry tachycardia, atrial tachycardia, ventricular tachycardias and premature ventricular ectopic beats.
There is a large range of antidysrhythmic drugs and in this module we will touch on only five in detail as they are examples of a particular class of antiarrhythmic drug and are the more commonly used antidysrhythmic agents.
Antidysrhythmic drugs are classified using the Vaughan Williams system based on their action on a particular phase of the cardiac action potential. There are other antidysrhythmic drugs that have come about since that system was first described. They fit into an ‘other’ category, so antidysrhythmic drugs are classified by their action using the following structure:
- Class 1a
- Class 1b
- Class 1c
- Class II
- Class III
- Class IV
All antidysrhythmic drugs have the potential to be pro-arrhythmic and so should be used with caution, particularly in higher doses.
Class 1a, 1b and 1c antidysrhythmic drugs
All class one drugs block voltage-sensitive sodium channels and interfere with the rapid influx of sodium into the cell in phase 0 of the cardiac cycle. They typically bind to the sodium channel when it is open or in the refractory phase.
There are differences between the drugs in this class (hence the sub-classes) but these are not so important for the purpose of this module. What is important is they all affect sodium influx (by altering the fast sodium channel) in phase 0 of the cardiac cycle (see Figure 1). Class 1 antidysrhythmics include quinidine, procainamide, disopyramide, lidnocaine, mexiletine and flecinide.
Class II drugs include atenolol, esmolol and metoprolol (discussed earlier).
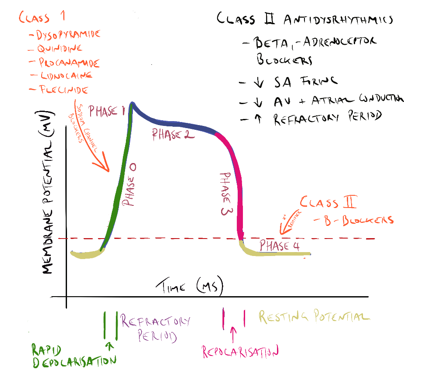 |
Class III antidysrhythmic drugs
Class III antidysrhythmic agents include amiodarone and sotolol. These drugs block the potassium channels in phase 3 of the cardiac cycle, effectively prolonging the action potential and increasing the refractory period of the cardiac cycle in the AV node and atrial and ventricular tissue.
- Amiodarone. Amiodarone is a class III antidysrhythmic that blocks potassium channels in phase 3 of the cardiac action potential and also has some effect on the sodium channels in phase 0. Amiodarone is used in the treatment of serious tachyarrhythmia’s including ventricular tachycardia AF and Supraventricular tachycardia (SVT). Amiodarone has a very long half-life. It can cause thyroid dysfunction, pulmonary toxicity and reversible corneal micro deposits. Therapeutic drug monitoring can identify compliance and toxicity but is rarely required. Amiodarone can also expose patients to skin photosensitivity manifesting in severe sunburn with limited sun exposure.
- Sotolol is a non-selective beta-blocker that prolongs the refractory period of the atria and ventricles and bypass tract. Sotalol also blocks the potassium channels in phase 3 of the cardiac action potential, again prolonging the action potential and increasing the effective refractory period in atrial and ventricular tissue and the AV node.
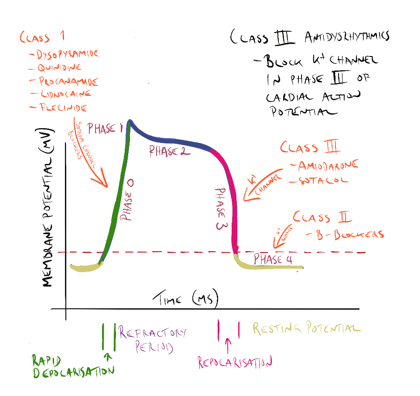 |
Class IV antidysrhythmic drugs
Class IV antidysrhythmic agents include the calcium channel blockers verapamil and diltiazem.
These drugs have been previously discussed
 |
📺 Watch the brief vodcast on Antiarrhythmics from Class I to Class IV (20 minutes)
Other antidysrhythmic agents
Atropine, adenosine and digoxin are used in arrhythmias. Digoxin is the final antidysrhythmic agent to be briefly examined.
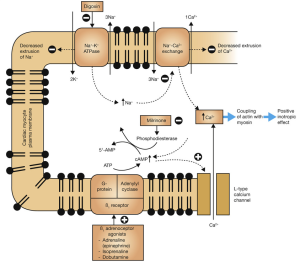
Digoxin is a cardiac glycoside used in atrial fibrillation, atrial flutter, and atrial tachycardia’s and sometimes in heart failure. Digoxin slows ventricular rate and also increases contractility of the heart. Digoxin is an inhibitor of the sodium/potassium pump in the myocardial cell. This pump is responsible for expelling intracellular sodium from the cell during repolarization – returning the cell to its original pre-depolarisation state. The binding of digoxin to the NA+/K+ ATPase inhibits its action. Elevated intracellular sodium levels prevent the removal of calcium ions making more calcium available for uptake by the sarcoplasmic reticulum (See figure below). The blockade of the NA+/K+ ATPase increases the refractory period in the atrial and AV nodal tissue (slowing heart rate) and the increased amount of calcium available to initiate actin-myosin binding increases the force of contraction.
- Digoxin has a narrow therapeutic index and digoxin toxicity can be present within the normal therapeutic range for the drug. It is common for patients taking digoxin to be monitored for signs of toxicity or undergo therapeutic drug monitoring.
 |
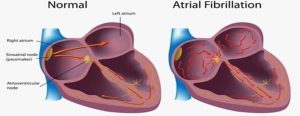
Atrial Fibrillation
Atrial fibrillation (AF) can be defined as “a supraventricular tachyarrhythmia with uncoordinated atrial electrical activation and consequently ineffective atrial contraction”
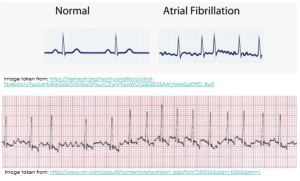
- When the atria contract and twitch randomly and out of rhythm, sometimes the P wave on the ECG is not discernible at all. Other times, like you can see in the diagram below, it appears as though there are multiple P waves before any given QRS complex (you will remember that the QRS complex indicates ventricular depolarisation).
 |
 |
Types of Atrial Fibrillation (AF)
When considering the different types of atrial fibrillation, as pharmacists, it is most important that we know whether the patient has valvular atrial fibrillation OR non-valvular atrial fibrillation.
Valvular atrial fibrillation
- Atrial fibrillation affecting a patient with moderate to severe mitral stenosis or a prosthetic heart valve
- Necessitates therapeutic anticoagulation with warfarin (see later)
Non-valvular atrial fibrillation
- Atrial fibrillation that is not associated with moderate to severe mitral stenosis or a prosthetic heart valve
Clinical Manifestation
- AF is commonly asymptomatic. It has been estimated that 1.4% of patients in our communities aged 65 years or more will have unknown/undiagnosed, asymptomatic AF at a single point in time random screening of their ECG trace.
- For patients who do experience symptoms associated with their AF, examples of such symptoms include palpitations, dyspnoea, symptomatic hypotension (e.g., dizziness, fatigue, confusion), and/or chest pain or discomfort.
Epidemiology
AF is the most common recurrent arrhythmia encountered in clinical practice. The prevalence of AF in Australia is estimated to be 2-4% but this is likely to be an underestimation since AF commonly exists undiagnosed.
Complications of AF
Thromboembolic consequences of AF are a cause of substantial morbidity and mortality. These include embolic stroke and pulmonary embolism. When the heart is not pumping blood in a coordinated manner from the atria to the ventricles, there is an increased chance of blood pooling and subsequently clotting within the heart.
- If a clot is ejected from the left side of the heart, this can cause an embolic stroke.
- If a clot is ejected from the right side of the heart, this can cause a pulmonary embolism.
Other possible complications of AF include myocardial infarction and heart failure.
📺 Watch final vodcast on arrhythmias and antiarrhythmics (15 minutes)
Summary
In this topic you have begun developing your knowledge around antiarrythmic drugs and you should now be able to:
- Identify and explain the different classifications of the antiarrthymic agents
- Explain the different actions on a particular part of the cardiac action potential of each class
- Identify and describe drugs that do not fit into any of those classes neatly and are classed as ‘other’.
COMMONWEALTH OF AUSTRALIA
Copyright Regulations 1969 WARNING This material has been reproduced and communicated to you by or on behalf of James Cook University in accordance with section 113P of the Copyright Act 1969 (Act). The material in this communication may be subject to copyright under the Act. Any further reproduction or communication of this material by you may be the subject of copyright protection under the Act. Do not remove this notice.