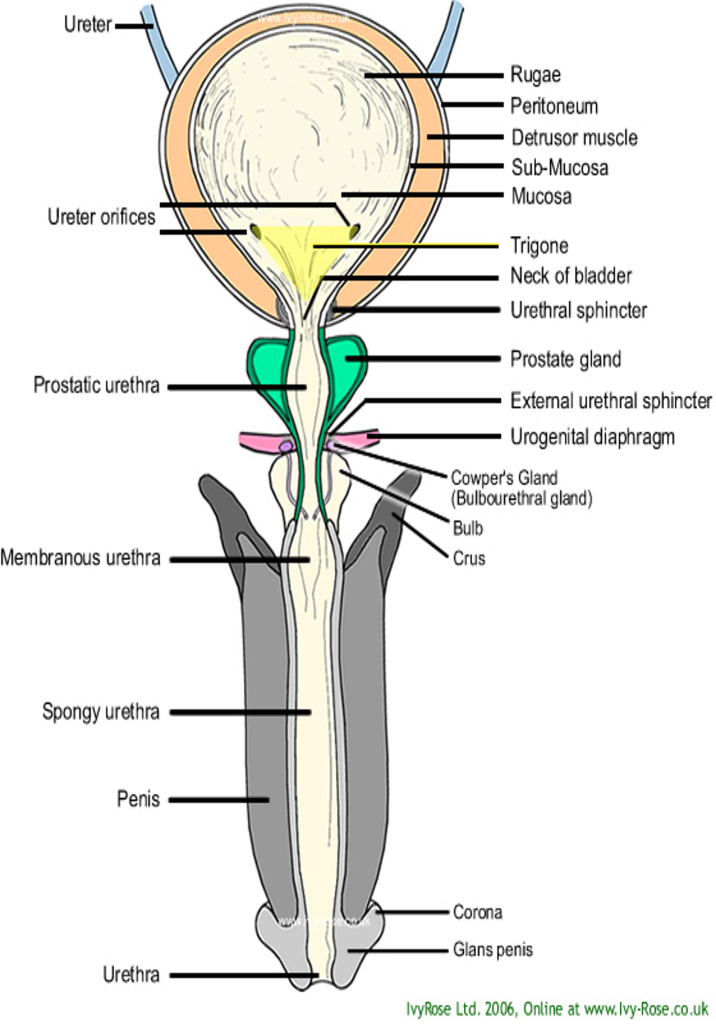
7.3 Genitourinary Drugs
Genitourinary Drugs
This topic will discuss some selective drugs that are used in different genital and urological disorders. The discussion will include the following:
- Erectile Dysfunction
- Urinary Incontinence and
- Benign Prostate Hyperplasia
1. Erectile Dysfunction
Erectile dysfunction (ED) is the inability to achieve or maintain a penile erection sufficient for satisfactory sexual performance. It is a relatively common ailment affecting men more as they advance in age. There are many causes of ED. They include:
- Advanced age,
- Atherosclerosis-related risk factors,
- Obesity and metabolic syndrome,
- Penile abnormalities,
- A range of commonly prescribed drugs and
- alcohol and illicit substances.
Many men do not seek treatment for ED due to a range of social, medical, financial and personal reasons.
Treatment of Erectile Dysfunction
There are 3 main treatment approaches to the drug management of erectile dysfunction.
- Phosphodisterase-5 inhibitors (PDE-5 inhibitors) that increase the amount of cGMP in the penis, decreasing intracellular calcium in the smooth muscle of the blood vessels causing engorgement.
- Use of injectable prostaglandin E1 analogue which causes the vasodilation of the blood vessels in the penis causing engorgement.
- Papaverine – a relaxer of the vascular components of penile erectile system.
The PDE-5 inhibitors are not the gold standard for the treatment and management of ED – that belongs to the injectable agent alprostadil. while PDE-5 inhibitors are not the gold standard, they are the most commonly used agent due to their convenience. The examples of PDE-5 inhibitors are sildenafil, tadalafil and avanafil.
Penile erection results from dilation of the corpus cavernosa smooth muscle and increased blood flow. The dilation occurs due to nitric oxide (NO), which stimulates soluble GC to synthesise cGMP, in turn activates PKG, leads to decreased cytosolic Ca2+ and smooth muscle relaxation. The enzyme, phosphodiesterase-5 normally breaks down cGMP.
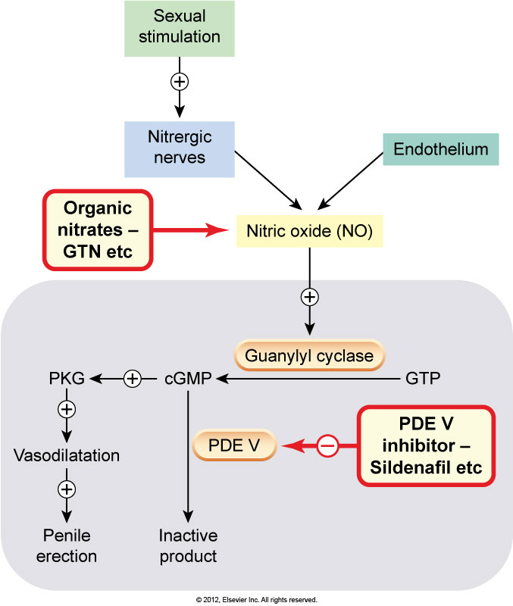
The PDE-5 inhibitors reduce the breakdown of cyclic GMP. elevated levels of cGMP decrease the influx of calcium into the smooth muscle of the arterioles and arteries, increasing blood supply to the corpus cavernosum. The resulting enlargement of the corpus cavernosum compresses venous outflow – both actions contributing to engorgement of the penis and erection.
Their onset of action is relatively short (15-60 minutes) and they can last for between 6-8 hours up to 36 hours depending on the agent. This does not mean the erection will last this long, but because the action of the PDE-5 inhibitors requires sexual stimulation, an erection can be achieved during this time.
The adverse effects of PDE-5 inhibitors are mostly dose related. Some adverse effects include headache (>10%), flushing, priapism, dizziness, dyspepsia, rhinitis, and vision loss due to non-arteritic anterior ischemic optic neuropathy (NAION).
These drugs are contraindicated with nitric oxide donors, organic nitrates or nitrites (including glyceryl trinitrate in any form, isosorbide mononitrate, isosorbide dinitrate, sodium nitroprusside, amyl nitrite, nicorandil). If a nitrate is required after a PDE5 inhibitor has been taken, wait at least 24 hours with sildenafil, 12 hours with avanafil and 48 hours with tadalafil before giving the nitrate. A longer period may be required if the elimination of the PDE5 inhibitor is delayed (eg due to drug interaction, renal or hepatic impairment). Patients with cardiovascular diseases should avoid using these drugs or precautions must be considered if desired to use these.
The 2 main other pharmacological treatments for ED are alprostadil and papaverine. Alprostadil is an injectable prostaglandin E1 analogue. PG-E1 is a potent vasodilator, causing local vasodilation of the cavernous artery and associated arterioles causing filling of the corpus cavernosum in much the same way as the PDE-5 inhibitors. These drugs are injected into the corpus cavernosum prior to intercourse causing dilation of the blood vessels, compression of the venous outflow and subsequent erection.
2. Urinary Incontinence (UI)
Lower urinary tract (LUT) consists of bladder, urethra, and sphincter muscles. Bladder is made up of 4 layers, serosa, detrusor muscle, submucosa and mucosa. Urine storage results when detrusor muscle relaxes whereas micturition occurs when it contracts.
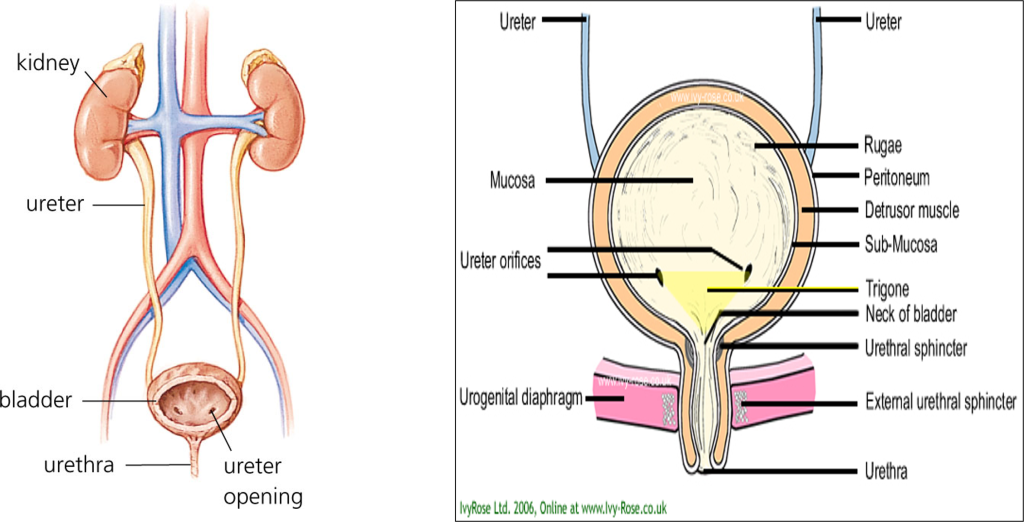
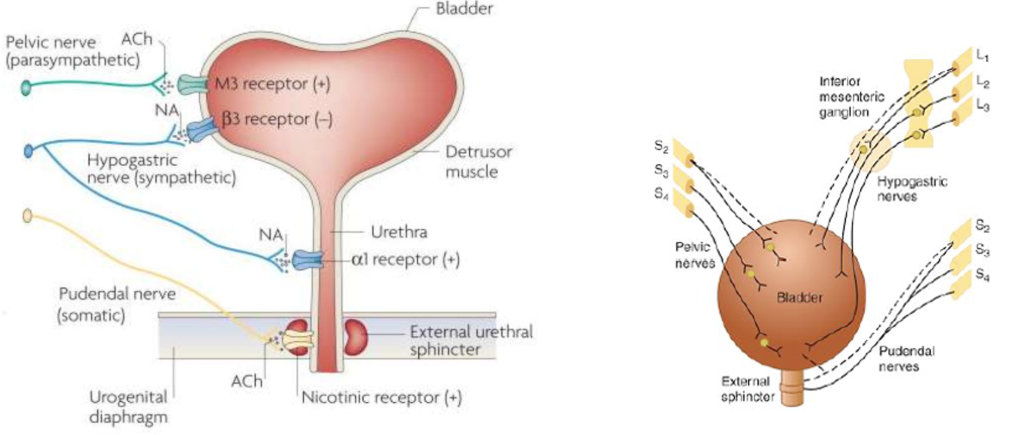
Contraction is brought about by the parasympathetic activity mediated by acetylcholine on muscarinic receptors (M3). Internal sphincter leading to the urethra is usually closed due to sympathetic activity mediated by noradrenaline. So, in case of micturition, sympathetic activity decreases and parasympathetic activity increases. These lead to relaxation of the internal sphincter and contraction of the detrusor muscle. Physiologically, UI results when the pressure of the bladder exceeds that of the urethra resulting in involuntary micturition.
Urinary incontinence is a common and embarrassing problem, especially among older adults. Urinary incontinence (UI) is a disorder of the reservoir function of the lower urinary tract and is defined as “the involuntary loss of urine which is objectively demonstrable, and which has become a social or hygienic problem”. UI is a condition often accompanied by other distressing lower urinary tract symptoms like urgency, frequent urination during the day, and nocturnal urination. Although UI is prevalent in both men and women, it remains a frequently underreported issue that can significantly affect an individual’s quality of life. Those who experience UI may feel a loss of self-control, independence, and self-esteem, leading them to alter their daily activities to avoid potential accidents. In addition to these psychological and social impacts, UI can also result in physical complications such as perineal skin irritation, infections, pressure sores, urinary tract infections (UTIs), and an increased risk of falls.
Before considering pharmacological interventions, it’s crucial to rule out any contributing factors. These may include urinary tract infections, excessive fluid consumption, metabolic issues like hyperglycemia, or the use of specific medications such as beta blockers, anticholinergics, antipsychotics and diuretics (see the table below).

Incontinence can be classified into several types, including:
- Overactive Bladder (Detrusor Overactivity): This type is marked by a sudden, intense need to urinate that is difficult to control, increased frequency of urination (more than eight times a day), and nocturia, which affects approximately 50% of men and women with overactive bladder (OAB) symptoms. These symptoms may occur with or without urge incontinence, which is the involuntary leakage of urine accompanied by an urgent need to urinate. The severity of OAB tends to increase with age, particularly after the age of 60, and can significantly affect the quality of life.
- Stress Incontinence: This occurs when the body fails to prevent urine leakage in response to an increase in abdominal pressure, such as during coughing or physical exertion.
- Overflow Incontinence: This type is caused by a failure to empty the bladder properly, leading to urine retention and bladder distension. It may result from an obstruction of the urinary outlet, such as in cases of prostatic hyperplasia, or from a neurogenic bladder, where the detrusor muscle is unable to contract effectively.
However, the International Continence Society has subdivided lower urinary tract symptoms into three groups:
- Storage symptoms that include increased daytime frequency, nocturia, urgency and urinary incontinence
- Voiding symptoms that include splitting or spraying, slow or intermittent stream, hesitancy, straining and terminal dribble
- Post-micturition symptoms include a sense of incomplete emptying, and post-micturition dribble.
Treatment/Management of Urinary Incontinence
There are several effective non-pharmacological management of UI available. These approaches include lifestyle/behavioural modifications, fluid management, bladder training (scheduled voiding regimens), pelvic floor muscle rehabilitation/exercise, external neuromodulation, anti-incontinence devices, acupuncture, and supportive interventions.
The pharmacological treatments of UI vary depending on the type of UIs. The common drugs used in UI are anticholinergics and desmopressin.
Anticholinergics – Oxybutynin and Solifenacin
Uroselective muscarinic antagonists (e.g., oxybutynin) are the first-line pharmacologic treatment for urge UI.
Oxybutynin is a competitive antagonist at muscarinic M1 and M3 receptors and has been utilized for over three decades in the management of overactive bladder (OAB). Besides its antimuscarinic activity, oxybutynin also exerts a direct antispasmodic effect on the detrusor muscle and possesses local anesthetic properties. However, these additional effects are significantly weaker—approximately 500 times less potent—than its antimuscarinic actions.
This drug is a racemic mixture, containing equal parts (50:50) of R-oxybutynin and S-oxybutynin, with the R-isomer being primarily responsible for its pharmacological effects. Oxybutynin undergoes metabolism in both the intestinal wall and liver, primarily through the enzyme CYP3A4, producing N-desethyloxybutynin (DEO) as the main metabolite. The drug’s absolute bioavailability is approximately 60%, and DEO plasma levels are typically 5 to 12 times higher than those of the parent compound. It’s suggested that DEO may contribute significantly to the anticholinergic side effects associated with oxybutynin. Only a minimal amount, less than 1%, is excreted unchanged. The time to reach peak drug concentration varies depending on the formulation, taking about an hour with the tablet form and 24 to 48 hours with the transdermal patch. The half-life also differs: it is 2 to 3 hours for the tablet and patch, while it extends to 12 to 13 hours for the extended-release formulation.
The adverse effects of oxybutynin include dry mouth, constipation, somnolence, dizziness, agitation, hallucinations and memory impairment Oxybutynin is best avoided in combination with other anticholinergic drugs (additive effects), people with dementia (it may worsen symptoms) and situations of pre-existing urinary retention or significant bladder outlet obstruction (it may worsen).
Desmopressin
Nocturia can result from a variety of factors, including uncontrolled diabetes mellitus, congestive heart failure, bladder or prostate disorders, and the use of certain medications, particularly diuretics. Nocturnal enuresis in children aged 10 years or more may warrant treatment with desmopressin (an analogue of ADH, given by mouth or by nasal spray for the treatment of diabetes insipidus caused by ADH deficiency due to disease of the posterior pituitary gland), combined with restricting fluid intake in the evening. Desmopressin is a synthetic analog of vasopressin is approved for the treatment of nocturia caused by nocturnal polyuria in adults who wake up at least twice per night to urinate. However, the use of this product is limited by several contraindications, particularly in older adults. These contraindications include hyponatremia, excessive fluid intake, primary nocturnal enuresis, concurrent use of loop diuretics or systemic/inhaled glucocorticoids, renal impairment, syndrome of inappropriate ADH secretion, coexisting conditions that may cause fluid or electrolyte imbalances, moderate to severe heart failure, and uncontrolled hypertension. The approval of intranasal desmopressin was based on clinical trials involving patients aged 50 and older.
📺 Watch the brief vodcast on sexual dysfunction and urinary incontinence (17 minutes)
3. Benign Prostate Hyperplasia (BPH)
The prostate is an organ, which is of the shape and size of a horse chestnut, that encircles the portion of the proximal posterior urethra that is located at the base of the urinary bladder. The prostate produces secretions, which are part of the ejaculate.
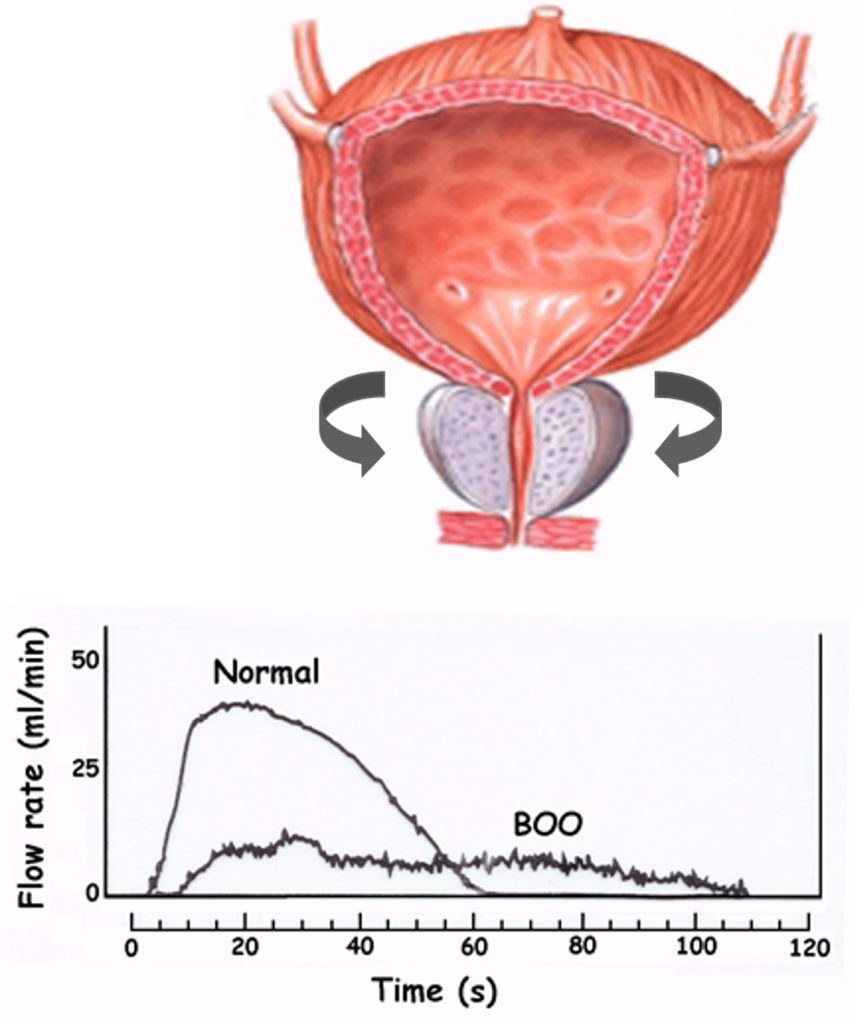
Benign prostatic hyperplasia (BPH) is the most common benign neoplasm in men who are at least 40 years of age. BPH can produce lower urinary tract symptoms (LUTS), a collection of obstructive and irritative voiding symptoms that are consistent with impaired emptying of urine from and defective storage of urine in the bladder, respectively. Medications are a common mode of treatment to reduce symptoms and/or delay complications of BPH.
BPH first presents as benign prostatic hyperplasia, a histologic disease in many elderly men that increases in prevalence with advancing patient age. Benign prostatic hyperplasia may progress to benign prostatic hypertrophy, which is clinically evident, and then to benign prostatic obstruction, which produces LUTS.
The testes and adrenal glands produce 90% and 10%, respectively, of circulating testosterone. Testosterone enters prostate cells, where predominantly Type II 5α-reductase converts testosterone to dihydrotestosterone, which combines with a cytoplasmic receptor. The complex enters the nucleus and induces changes in protein synthesis that promote glandular tissue growth of the prostate.
The prostate is composed of two types of tissue: (a) glandular or epithelial tissue, which produces prostatic secretions, including prostate-specific antigen (PSA), and (b) muscle or stromal tissue, which can contract around the urethra and bladder outlet when stimulated. Whereas androgens stimulate glandular tissue growth, they have no direct effect on stromal tissue. Stromal tissue growth may be stimulated by estrogen. Because testosterone is converted to estrogen in peripheral tissues in males, testosterone may be associated indirectly with stromal hyperplasia. Stromal tissue is innervated by α1A-adrenergic receptors. When stimulated, prostatic stroma contracts around the urethra, narrowing the urethra and causing obstructive voiding symptoms.
Treatment of BPH
Until recently, the principal approach to treatment focused on reducing BPH symptoms. However, treatment should also slow disease progression and decrease complications of BPH, particularly in those patients at high risk
5α-reductase inhibitors
5α-reductase inhibitors act by a very different mechanism: they specifically inhibit 5α-reductase enzyme type II, which in the prostate gland metabolises the conversion of testosterone to DHT, a more potent androgen responsible for prostate gland growth. Examples of this class include dutasteride and finasteride. These drugs reduce levels of DHT in the bloodstream and in the prostate, reducing hypertrophy of the gland and resistance to urinary outflow. They do not impair synthesis of testosterone and so do not have anti-androgenic actions. They appear most effective in men with large prostates and may take 6 months of treatment for clinical improvement.
Dutasteride, with chronic administration, reduces prostate size, alleviates symptoms of BPH, improves urinary flow and reduces the need for surgery. It is indicated for mild to moderate symptoms of BPH with clinically demonstrated prostatomegaly, when surgical treatment is contraindicated or refused. It may be co-administered with an α-blocker such as tamsulosin. The adverse effects of dutasteride include decreased libido, impotence, decreased sperm count and amount of ejaculate, gynaecomastia and allergic reactions. There is a possible association with increased risk of male breast cancer or prostate cancer, so changes in breast tissue or signs of prostate cancer should be monitored. When co-administered with an α-blocker, combined adverse effects include hypotension and syncope.
Alpha 1 receptor antagonists
Stimulation of the α1-adrenoceptors in the bladder neck promotes an increase in smooth muscle tone of the bladder neck, which increases bladder outlet resistance. Therefore, the use of α1-adrenoceptor antagonists (e.g., tamsulosin and prazosin) results in relaxation of the smooth muscle of the bladder neck, thus decreasing muscle tone. As a consequence, urethral pressure decreases, bladder outlet resistance is reduced and the obstruction to urine outflow is lessened. These are indicated for symptoms relief in benign prostate hyperplasia.
All α1-adrenergic antagonists are considered equally effective in relieving symptoms. The adverse effects include orthostatic hypotension, dizziness, retrograde ejaculation, impotence.
📺 Watch the brief vodcast on benign prostate hyperplasia, BPH (11 minutes)
Lecture Notes
MD2012_W7_Lecture 3_Genitourinary
COMMONWEALTH OF AUSTRALIA Copyright Regulations 1969 WARNING
This material has been reproduced and communicated to you by or on behalf of James Cook University in accordance with section 113P of the Copyright Act 1969 (Act).
The material in this communication may be subject to copyright under the Act. Any further reproduction or communication of this material by you may be the subject of copyright protection under the Act. Do not remove this notice.