Want to create or adapt books like this? Learn more about how Pressbooks supports open publishing practices.
1.3.2 Pharmacotherapy for Stable Angina
Pharmacotherapy for Stable Angina
Organic Nitrates
The organic nitrates are effective dilators of (predominantly) veins and arteries and are used to treat angina pectoris. Organic nitrates can be classed into two broad groups:
Short acting rapid onset nitrates: glyceryl trinitrate and isosorbide dinitrate
Short and long-acting nitrates are both used to manage angina but are used at different times. The short acting – rapid onset sublingual glyceryl trinitrate and isosorbide dinitrate are used to manage acute symptoms of unstable angina or for the prevention of symptoms of angina. The longer acting nitrates isosorbide mononitrate (and controlled release isosorbide dinitrate and glyceryl trinitrate patch) are used to prevent symptoms of chronic angina. The order of relative duration of action of the different nitrates can be remembered by the number of nitrate groups in the preparation:
Trinitrate (3) is short acting
Dinitrates (2) slightly longer acting
Mononitrates (1) long acting.
Endogenous (naturally produced) nitric oxide is produced by vascular endothelial cells (other cells too but they are less important at the moment) and has a range of important functions. One of these functions is to mediate relaxation of vascular smooth muscle. Nitric oxide (NO) moves from the endothelial cell and enters the smooth muscle cell where it binds with soluble guanylyl cyclase which converts GTP to cGMP. cGMP activates Protein Kinase G which phosphorylates myosin and causes smooth muscle relaxation.
📺 Watch the YouTube video on the role of NO in smooth muscle relaxation. Video available at (3:30 min)
The organic nitrates donate a nitrate group where it is converted to nitric oxide by s-nitrosothiol to nitric oxide. Nitric oxide (NO) binds with soluble guanylyl cyclase which converts GTP to cGMP. cGMP activates Protein Kinase G which phosphorylates myosin and causes smooth muscle relaxation.
Action of exogenous nitrates – dontates a NO group which is a cofactor for the conversion of GTP to cGMP. Increased levels of cGMP cause relaxation of vascular smooth muscle. Image taken from Klabunde, R. General Pharmacology. National Library of Medicine (US); 2007 [updated 2012 July 11; cited 2020 March 7]. Available at: https://cvpharmacology.com/vasodilator/nitro
At therapeutic doses of nitric oxide, the dilation of veins predominates over arteries. The vasodilatation of veins (+++ lots) and arteries (+ a bit) has 2 major effects
It is the reduction in ventricular pre-load and resultant reduction of myocardial oxygen demands that explains its benefit in stable angina (pectoris), rather than it’s dilation of the coronary arteries. Side effects include orthostatic hypotension, headache, flushing, palpitations, hypotension and peripheral oedema.
Patients can build a tolerance to nitrates with continuous exposure. This can be avoided by ensuring patients have nitrate-free period of at least 8-hours in each 24-hour period Kinetics.
Most organic nitrates have low oral bioavailability as they undergo extensive first pass metabolism. The one exception is isosorbide mononitrate which has quite high oral bioavailability. Because the bioavailability of the other organic nitrates are low, they are typically administered by sublingual tablet or spray, patch or infusion.
📺 Watch the vodcast on the Pharmacology of nitrates. (6:03 minutes)
Summary
The organic nitrates donate a nitrate group providing an exogenous (artificial) source of nitric oxide to cause vasodilatation.
This results in a reduction of available cytosolic calcium and decreased venous (and arterial to a lesser extent) contraction.
This results in a decrease in preload and therefore myocardial oxygen demand, rebalancing the supply-demand mismatch that explains angina.
Organic nitrates can be grouped into short and long-acting classes.
The useful effect in angina comes from the reduction in pre-load rather than the physical dilatation of the coronary arteries.
Beta-Adrenoceptor Antagonists (aka Beta-blockers)
Beta-adrenoreceptor antagonists or beta-blockers competitively block the effect of catecholamines (noradrenaline and adrenaline) at the beta-adrenoceptors. While beta receptors are widely distributed throughout the body, beta2 receptors are found mainly in the smooth muscle of the airways and beta1 receptors are located in large populations in myocardial tissue. When the sympathetic nervous system releases noradrenaline or adrenaline, the beta-antagonist blocks the effect of the catecholamine thus reducing heart rate and contractility associated with beta receptor stimulation.
How does the beta antagonist work at a cellular level?
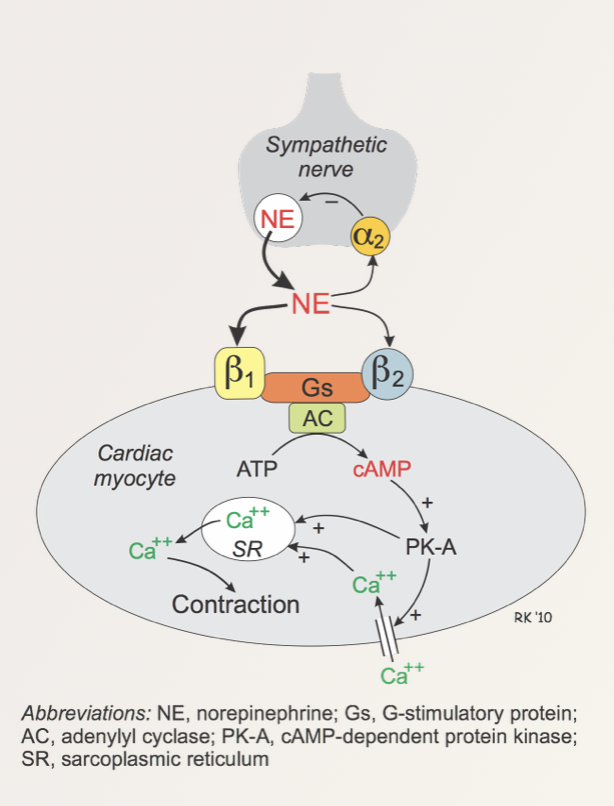
The beta1 receptor is a Gs-coupled protein receptor as seen below in the figure below.
The effect of stimulation of the beta receptors on cardiac myocyte contraction. Source: Klabunde, R. CV Pharmacology. All rights reserved
The binding or noradrenaline (or adrenaline) to the beta1 receptor on the cardiac myocyte under normal conditions activates adenylyl cyclase which promotes the conversion of ATP to cAMP. Increased levels of cAMP activates cAMP-dependent protein kinase (PK-A). PK-A opens L-type calcium channels increasing the amount of calcium (Ca2+) in the cardiac monocyte as well as encouraging the release of calcium from the sarcoplasmic reticulum. The increased intracellular calcium binds to cardiac troponin-C component of the troponin complex on the actin thin filaments on the cardiac muscle.
This moves tropomyosin out of the way allowing the myosin head to interact with the actin facilitating contraction. In short, increased intracellular calcium allows more myosin heads to interact with the actin filament on the cardiac muscle – this allows an increased contraction of the muscle tissue (see video explanation below).
Beta-antagonists block the action of the catecholamines at the beta-receptor thereby decreasing the amount of intracellular calcium that is available and this in turn reduces contraction of the heart.
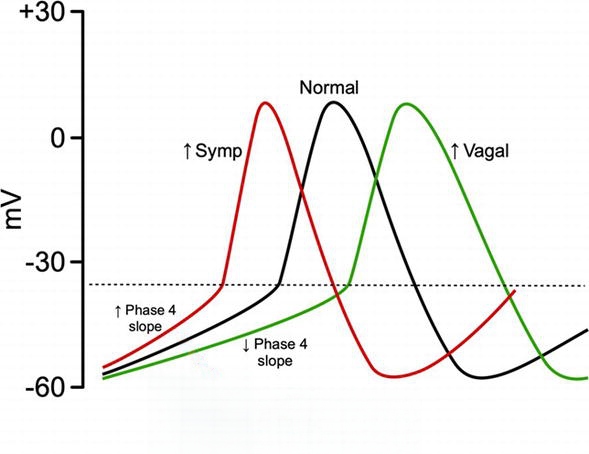
The effect of the beta-receptor on heart rate is also linked to intracellular calcium stores. Stimulation of the beta-receptors in the SA node, AV node and atrial tissue increase the intracellular concentration of calcium in a similar fashion to that described previously – it facilitates the opening of L-type calcium channels allowing the movement of calcium into the cell and encourages the release of calcium from the sarcoplasmic reticulum. The increase in intracellular calcium levels alters the electrical potential of the cell bringing it closer to threshold (about -40 to -30 mV) where phase 0 of the action potential (depolarization) can occur (see figure below).
Action potential of nodal tissue in the heart. Note there is no resting membrane potential. The membrane potential continuously moves in a more positive direction from the completion of the previous cycle. Source: CV Physiology. All rights reserved
📺 Watch the YouTube video on how calcium increases contraction of the cardiac myocyte.
There are three main groups of the beta-adrenoreceptor antagonists:
B1-selective blockers or cardioselective blockers because they block Beta1-adrenoceptors mainly located in the heart.
Non-selective B-adrenoreceptor antagonists which block both types of adrenoreceptors B1 and B2, and
Non-selective B-adrenoreceptor antagonists with intrinsic sympathomimetic activity (ISA) which causes partial stimulation of the B-adrenoreceptor with the effect being less than that of a pure agonist.
The blockade of B1 receptors in myocardial tissues produces the following effects:
In the Sinoatrial node blockade reduces heart rate (negative chronotropic effect)
In the Myocardial tissue blockade reduces contractility (negative ionotropic effect)
In the Juxtaglomerular cells in the kidney blockade reduces the release of renin (important in the reabsorption of sodium in the distal tubule and collecting ducts)
In the Central nervous system blockade reduce outflow of the sympathetic nervous system from the medulla oblongata.
The reduction in heart rate and contractility combined with a reduction in the release of renin and a reduced outflow of the sympathetic nervous system reduces blood pressure – a very common use of beta-blockers.
The effect of beta-blockers on a resting heart is modest, it exerts a more significant reduction in heart rate and contractility during exercise, so beta-blockers also reduce exercise tolerance. The benefit of a reduction in heart rate and contractility aside from a reduction in blood pressure is it also reduces myocardial tissue oxygen demand, which makes it useful in people with conditions with reduced coronary artery blood flow (for example angina).
Listed below are the B-adrenoreceptor antagonist (Beta Blocker) drugs in their classifications:
Beta-blockers can also be classed according to their lipid solubility. The lipid soluble beta-blockers tend to have greater central nervous system adverse effects (they penetrate the CNS more readily) and are typically metabolized by the liver. Most beta-blockers are lipid soluble. Where unwanted central nervous system side effects occur with the use of beta-blockers, for example, nightmares, a beta-blocker with low lipid solubility such as atenolol or sotalol may be more appropriate.
Common adverse effects of beta blockers
There are some clinical conditions where beta blockers are contraindicated because of their adverse effects such as in asthma and chronic obstructive pulmonary disease (COPD). This is because Beta blockers can precipitate bronchospasm leading to airway compromise. Remember that the B2 receptor was found on the smooth muscle of the airways?
Other adverse effects include bradycardia, orthostatic hypotension, cold extremities, fatigue, changes in glucose and lipid metabolism and they can have a transient negative effect on symptoms of heart failure.
📺 Watch: The recorded video on beta-blockers. (31:36 min)
Summary
Selective beta blockers affect mainly Beta1 receptors in the myocardial tissue.
Non-selective beta-blockers interact with Beta1 receptors and Beta2 receptors accounting for some of the respiratory side effects
Beta-blockers can be stratified by beta-selectivity, lipid solubility and intrinsic sympathomimetic activity
Beta-blockers block a G-protein coupled receptor which ultimately reduces calcium in the cell reducing automaticity (slowing heart rate) and decreasing contractility.
Beta-blockers have a modest effect at rest – they have a more significant effect during exertion.
📚 Read/Explore
Read section 6.3.6 Beta-blockers (Page 277-282) in Australian Medicines Handbook. Adelaide: Australian Medicines Handbook Pty Ltd; 2020
Calcium Channel Blockers
Calcium Channel Blockers (CCB’s) are commonly used in the management of hypertension and angina. They act on calcium channels in myocardial and vascular smooth muscle to:
Reduce heart rate and contractility, and
Reduce vasoconstriction of vascular smooth muscle.
The CCB’s available in Australia include amlodipine, clevidipine, diltiazem, felodipine, lercanidipine, nifedipine, nimodipine and verapamil. Calcium channel blockers are commonly grouped into two main classes:
Non-dihydropyridine calcium channel blockers
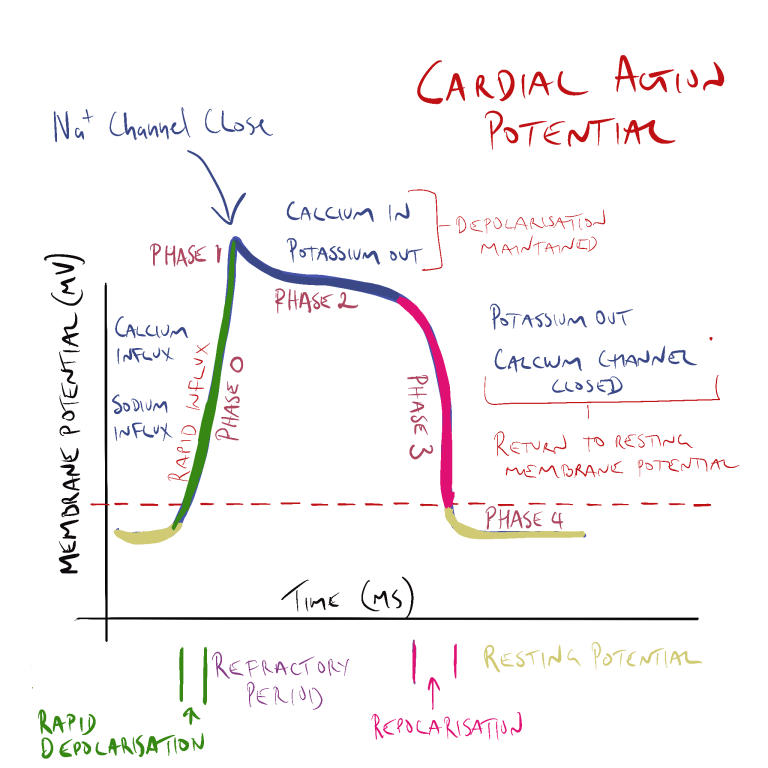
The non-dihydropyridine CCBs act mainly on cardiac and arteriolar smooth muscle to reduce contractility, heart rate, conduction and arteriolar vessel constriction. Contractility of the cardiac muscle cell is reduced as CCB’s block the inward movement of calcium through L-type calcium channels in phase 0 and phase 2 of the cardiac action potential. As there is less calcium entering the cell and less calcium is released from the sarcoplasmic reticulum. As less calcium is available to interact with troponin-C and less myocin heads are able to bind with the actin, contraction of the myocardial tissue is reduced.
In sinoatrial (SA) and atrioventricular (AV) junction node tissue, non-dihydropyridine CCB’s decrease automaticity by blocking the inward flow of calcium ions into the cell slowing depolarization in phase 0 of the cycle (remember –depolarization of nodal tissue is normally triggered by the inward flow of calcium into the cell – not sodium as in heart and skeletal muscle cells). Blocking the influx of calcium decreases the rate of depolarization depressing automaticity. This increases refractory time and decreases heart rate.
Dihydropyridine calcium channel blockers
These act mainly of arteriolar smooth muscle. The dihydropyridine CCBs share a common suffix ‘ipine’, (amlodipine, clevidipine, felodipine, lercanidipine, nifedipine and nimodipine) which makes them easy to spot in a crowd.
Like the non-dihydropyridine CCBs, they block the inward flow of calcium into the coronary arteries and arterioles in the peripheral circulation. The functional effect is the dilatation of arterioles and coronary arteries is to reduce peripheral vascular resistance and increase nutrient and oxygen supply to the heart. The following video briefly shows the effect of reduced calcium influx into the arteriolar smooth muscle cell and its negative effect on the activation of the myosin-actin complex.
📺 Watch: Mechanism of action for Calcium Channel Antagonists (0:44 minutes)
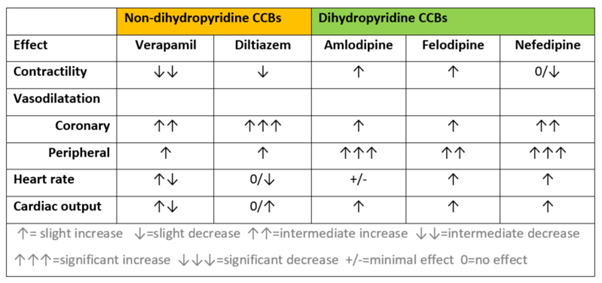
While the non-dihydropyridine CCB’s affect both arteriolar smooth muscle and cardiac nodal and cardiac muscle cells, they have a much more pronounced negative central cardiovascular effect. That is they have greater negative chronotropic and ionotropic effect compared with the dihydropyridine CCBs which act mainly on arteriolar smooth muscle. Of the two non-dihydropyridine CCBs, verapamil has the greatest effect on contractility and automaticity and cannot be used with other drugs that also reduce contractility or heart rate (such as the beta-blocking drugs). The table below provides a comparison of the cardiovascular effects of calcium channel-blocking drugs available in Australia.
From the table above, you can see the calcium channel blockers can have different effects depending on the agent and sub-class. This is because unlike many other drug classes – CCBs have fairly diverse chemical structures.
The trends worth noting are:
Non-dihydropyridine CCBs reduce contractility of the heart.
All produce some degree of coronary artery or peripheral artery vasodilatation.
They have mixed effect on heart rate.
They all trend towards increasing cardiac output.
Amlodipine has a long half-life and is typically given once a day. The other CCBs typically have a shorter active half-life and are either given more than once a day or are formulated in control release tablets. A large number of drugs interact with CCBs as they are primarily metabolized by CYP3A4 (which is affected by a number of drugs) (all undergo hepatic metabolism).
Probably the best-known drug interaction is between beta-adrenergic blocking drugs and verapamil. As both drugs reduce cardiac rate and contractility, the additive effect risks heart block and severe bradycardia. Common adverse effects include constipation, bradycardia, hypotension, dizziness, skin flushing and rash, dry mouth and oedema of the ankles. CCBs can also cause gingival hyperplasia (not heard of gingival hyperplasia before? Why not look it up on the web?).
📺 Watch the following short video explaining the pharmacology of calcium channel blockers. (20.59 min)
Secondary/Event Prevention of Stable Angina
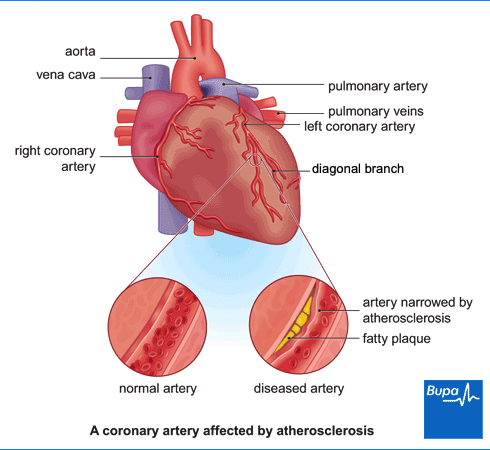
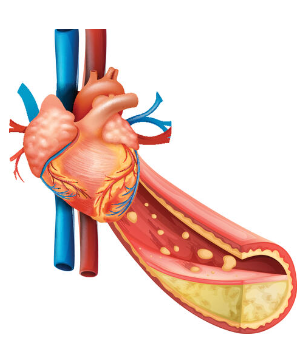
Managing patients with atherosclerotic cardiovascular disease (ASCVD), including those with stable angina, requires a comprehensive approach to secondary prevention. This strategy aims to prevent the recurrence of cardiovascular events and reduce overall mortality.
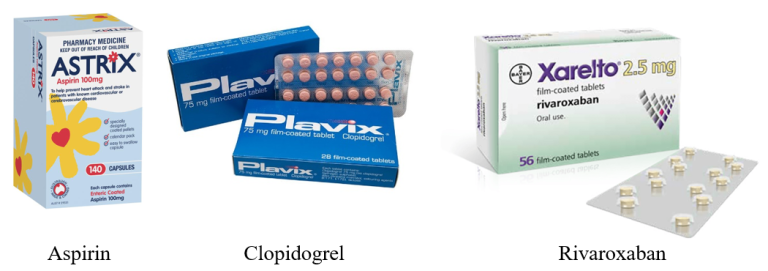
Secondary prevention typically involves a combination of medications aimed at reducing cholesterol, high blood pressure and reduce the risk of clot formation. Statins are commonly prescribed to lower cholesterol levels, stabilize atherosclerotic plaques, and in some cases, reduce the size of existing plaques. Antiplatelet drugs, such as aspirin or clopidogrel, or a combination of aspirin and rivaroxaban, are used to prevent thrombosis, thereby reducing the incidence of myocardial infarction and death.
The two most commonly used antiplatelet drugs for the prevention of platelet plugs around atherosclerotic plaques are aspirin or clopidogrel (if aspirin is not tolerated). Less commonly, aspirin plus rivaroxaban is used.
Low-dose (100-150 mg) enteric-coated aspirin is a common, effective and cost-effective antiplatelet option. The tablet is enteric coated to reduce the risk of gastric irritation and is given at a low dose reducing the risk of other side effects. If patients cannot tolerate aspirin, clopidogrel (75 mg once daily) serves as a viable alternative. Rivaroxaban may be beneficial in patients with poly-vascular disease when added to aspirin, although it increases the risk of gastrointestinal bleeding.
The cost of antiplatelet drugs is an important consideration.
Aspirin is cost-effective, with a monthly supply priced at approximately $4.17, whereas clopidogrel costs about $18.80 per month, and rivaroxaban is significantly more expensive at around $60.59 per month.
Despite the costs, these medications are relatively inexpensive compared to the financial burden of a myocardial infarction.
Blood Pressure Management
Another critical aspect of secondary prevention is blood pressure management. Effective control of hypertension is essential in reducing cardiovascular risk. Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and dihydropyridine calcium channel blockers are typically recommended for managing high blood pressure. Reviewing the previous chapter on blood pressure management is important for optimizing treatment strategies.
In summary, secondary prevention of atherosclerotic cardiovascular events involves a multifaceted approach that includes both lifestyle modifications and pharmacological treatments. Implementing these strategies effectively can significantly reduce the risk of recurrent cardiovascular events and improve patient outcomes.
Mechanism of Action of Antiplatelet Drugs in Secondary Prevention of Atherosclerotic Cardiovascular Events
In the context of secondary prevention for atherosclerotic cardiovascular events, understanding the role of antiplatelet drugs is essential. These medications are designed to reduce the risk of blood clot formation by interfering with the platelet activation and aggregation processes, which are critical early steps in clot formation.
Platelet Adhesion and the Cascade of Events
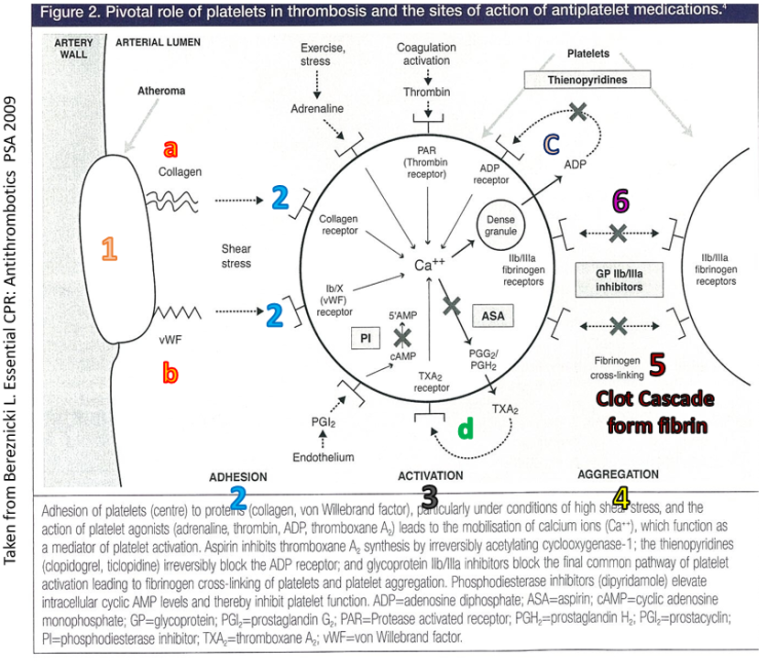
During blood clot formation, platelet adhesion is an early and crucial step. When atherosclerotic plaques in coronary arteries rupture or when there is significant turbulence due to narrowing of the vessel, it leads to the exposure of collagen and von Willebrand factor (vWF). This exposure triggers platelet adhesion and secretion, initiating the clotting cascade. Collagen and vWF act together as adhesive proteins that bind platelets to the site of vascular injury. They also activate the platelet to recruit other platelets.
Pivotal role of platelets in thrombosis and the sites of action of antiplatelet medications. Figure 2 from Bereznicki, L. Essential CPD: Antithrombotics. PSA 2009
Role of Thromboxane A2
Once platelets adhere to the damaged vessel wall, they produce thromboxane A2 under the influence of the enzyme cyclooxygenase-1 (COX-1). Thromboxane A2 plays a pivotal role in platelet aggregation and causes vasoconstriction by inducing contraction of vascular and respiratory smooth muscles. In the context of angina, thromboxane A2 contributes to the narrowing of the coronary artery, further exacerbating the condition by increasing platelet aggregation and formation of a platelet plug. It also causes the release of factors that recruit other platelets to the area of damage.
Aspirin’s Mechanism
Aspirin is a widely used antiplatelet drug that functions as a non-specific COX enzyme inhibitor. By inhibiting COX-1, aspirin effectively reduces the production of thromboxane A2 in platelets. This inhibition decreases platelet aggregation and minimizes vasoconstriction, thereby reducing the risk of clot formation in narrowed coronary arteries. The therapeutic use of aspirin is particularly beneficial in secondary prevention to prevent further thrombotic events in patients with existing coronary artery disease.
Clopidogrel’s Mechanism
Clopidogrel, another key antiplatelet agent, is a P2Y12 receptor antagonist. It works by blocking the action of adenosine diphosphate (ADP) at the P2Y12 receptor on platelets. This inhibition prevents ADP from activating platelets, thereby reducing their ability to aggregate. Clopidogrel is often used as an alternative to aspirin, especially in patients who are intolerant to aspirin or require additional antiplatelet therapy.
Both aspirin and clopidogrel play crucial roles in the secondary prevention of atherosclerotic cardiovascular events by targeting different mechanisms in the platelet activation and aggregation pathways. Aspirin inhibits COX-1 to reduce thromboxane A2 production, while clopidogrel blocks ADP-induced platelet activation. Understanding these mechanisms helps in effectively managing patients at risk of recurrent cardiovascular events.
📺 Watch the vodcast on the mechanism of action of antiplatelet drugs for secondary prevention in angina. (7:05 minutes)
Download the lecture notes here:
COMMONWEALTH OF AUSTRALIA Copyright Regulations 1969 WARNING
This material has been reproduced and communicated to you by or on behalf of James Cook University in accordance with section 113P of the Copyright Act 1969 (Act).
The material in this communication may be subject to copyright under the Act. Any further reproduction or communication of this material by you may be the subject of copyright protection under the Act. Do not remove this notice.

![Klabunde, R. General Pharmacology. National Library of Medicine (US); 2007 [updated 2012 July 11; cited 2020 March 7]. Available from: https://cvpharmacology.com/vasodilator/nitro.](https://jcu.pressbooks.pub/app/uploads/sites/119/2024/08/nitrodilator-mech.png)