5.1.1 Drugs That Affect Coagulation
Drugs That Affect Coagulation
Learning Outcomes
Be able to:
- Explain the cellular mechanism of action of the different classes of anticoagulants, thrombolytics and antiplatelet drugs
- Describe the major adverse effects, contraindications and important diet/drug interactions associated with anticoagulants, thrombolytics and antiplatelet drugs
- Explain the importance of drug monitoring when using warfarin, and outline the role of the INR in patient management
- Outline the therapeutic rationale for the use of these drugs in the treatment and prevention of acute coronary syndromes, stroke, transient ischemia attack and venous thromboembolism
- Name the other classes of drugs used in the general management of acute coronary syndrome (myocardial infarction and unstable angina).
 |
 |
 |
Introduction
Inappropriate activation of the coagulation pathways is linked to considerable morbidity and mortality. The risk of thrombosis increases with age, and each year, coronary heart disease and stroke claim thousands of lives. Additionally, numerous individuals are diagnosed with conditions such as deep vein thrombosis, pulmonary embolism, transient ischemic attacks, and other non-fatal thrombotic events. Thrombosis in an intact vessel typically arises from local trauma, vascular stasis, and systemic changes in blood coagulation.
There are three (3) major classes of drugs used to manage conditions associated with thrombosis. These are:
1. Anticoagulants (including Indirect Parenteral Anticoagulants, Direct Thrombin Inhibitors, Direct Factor Xa Inhibitors and Vitamin K Antagonists).
2. Antiplatelets.
3. Thrombolytics or Fibrinolytics.
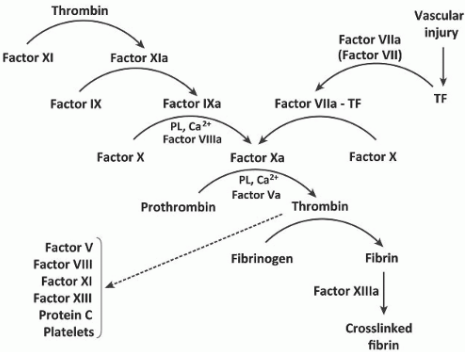
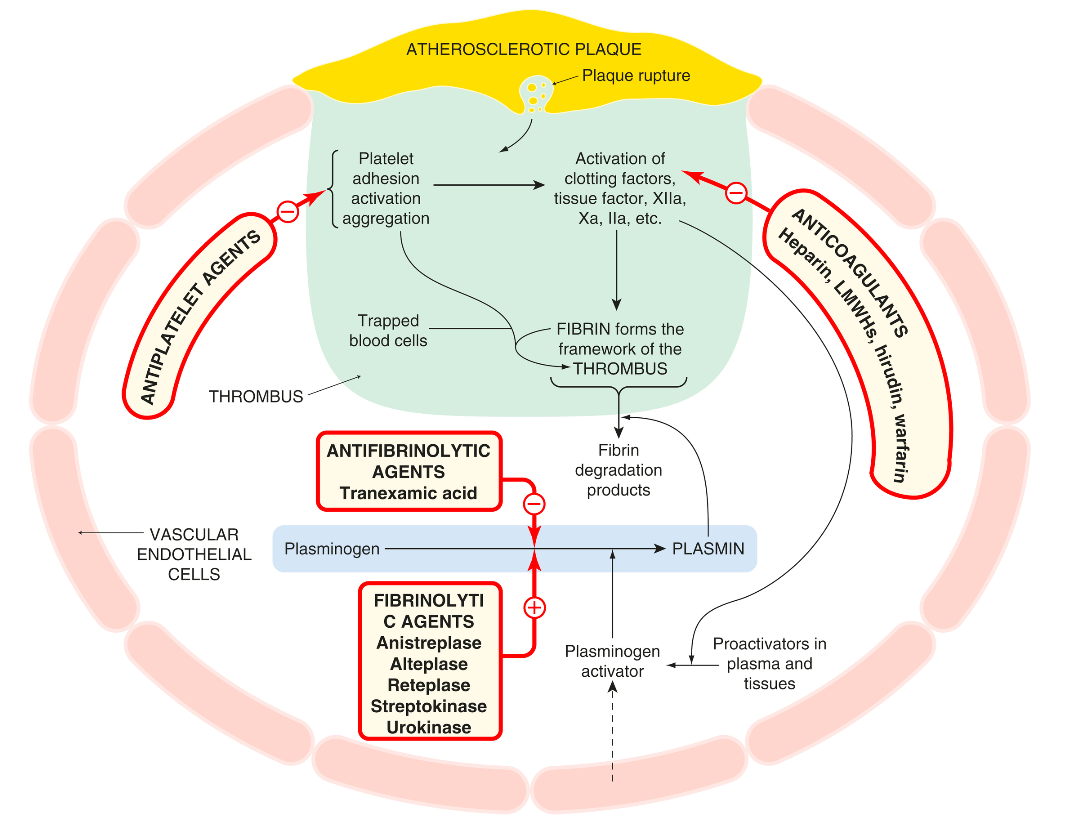
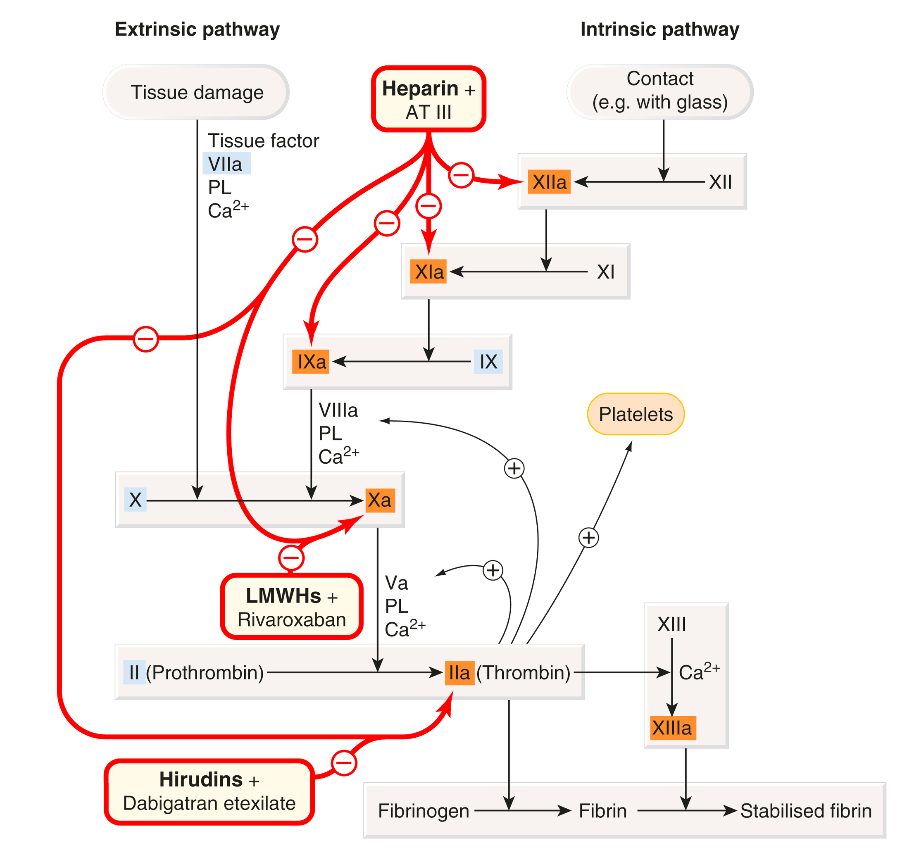
The figure below shows the sites of action of these drug classes interacting with the coagulation cascade and the fibrinolytic and platelet activation pathways.
 Watch the brief vodcast on the introduction of antithrombotic (11 minutes)
Watch the brief vodcast on the introduction of antithrombotic (11 minutes)
Anticoagulants
Anticoagulant medications are primarily used as prophylactics as they work by inhibiting the formation of fibrin deposits, preventing the growth of existing clots, and reducing the risk of thromboembolic events. However, it is crucial to understand that these medications do not have a direct impact on clots that have already formed or on tissue that has been damaged due to reduced blood flow caused by the clot. The main groups of anticoagulant drugs and drug agents in these classes include:
- Heparin and the low-molecular-weight heparins (LMWHs).
- Heparin.
- LMWHs = dalteparin, enoxaparin.
- LMW heparinoid = danaparoid.
- Direct thrombin inhibitors.
- Bivalirudin and dabigatran.
- Direct Factor Xa inhibitors.
- Apixaban and rivaroxaban.
- Antithrombin III-dependent anticoagulant = fondaparinux.
- Vitamin K antagonists.
- Warfarin.
As we discuss these agents in more detail it is important to know that direct thrombin inhibitors and direct factor Xa inhibitors are also known as direct oral anticoagulants (DOACs). These have mostly replaced the use of warfarin. It is also vital that you understand heparin and DOACs begin to work immediately, while warfarin requires several days to become effective.
Heparin
In 1916, a second-year medical student at Johns Hopkins Hospital discovered heparin while trying to isolate coagulant substances from various tissues during a vacation project. Instead of finding a coagulant, he identified a potent anticoagulant, which was later named heparin because it was initially derived from the liver. Commercial heparin is extracted from sources such as beef lung or pig intestine. As the different preparations vary in strength, they are biologically assayed against an international standard. Consequently, doses are measured in units of activity rather than by weight. Low-molecular-weight heparins (LMWHs) such as enoxaparin and dalteparin are preferred over unfractionated heparin (UFH) due to their longer duration of action. UFH is generally reserved for specific cases, such as in patients with renal failure where LMWHs are contraindicated. See the table below for overall comparison.

Both UHF and LMWH heparins are capable of inactivating factor Xa. UFH has the added ability to inactivate thrombin (IIa) by simultaneously binding to both antithrombin III and thrombin. In contrast, LMWHs primarily enhance antithrombin III’s activity against factor Xa. Due to their smaller molecular size, LMWHs cannot simultaneously bind to antithrombin III and thrombin, which results in a more pronounced inhibition of factor Xa and a relatively minor influence on aPTT. Danaparoid acts as an even more selective inhibitor of factor Xa compared to LMWHs. The schematic image below shows the interactions of heparins, antithrombin III (AT III) and clotting factors. To increase the inactivation of thrombin (IIa) by AT III, heparin needs to interact with both substances (top), but to speed up its effect on factor Xa it needs only to interact with AT III (middle). LMWH increase the action of AT III on factor Xa (bottom) but cannot increase the action of AT III on thrombin (IIa) because they cannot bind both simultaneously.
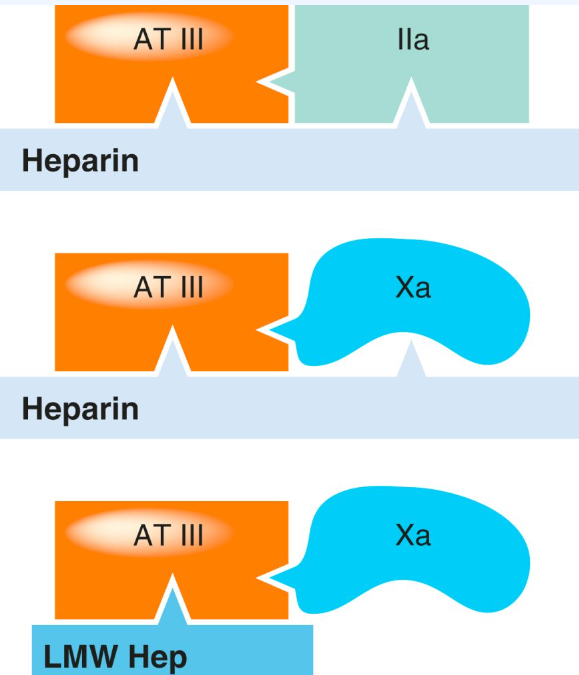
Heparin is not absorbed through the gastrointestinal tract due to its large molecular size and negative charge, necessitating its administration via intravenous or subcutaneous routes (intramuscular injections are avoided to prevent haematomas). Following an intravenous bolus injection, heparin undergoes rapid redistribution, followed by a slower elimination phase. This slower phase results from a combination of saturable processes, such as binding to endothelial cells and macrophages, and slower non-saturable processes, including renal excretion. Consequently, the plasma concentration of heparin increases more than proportionally with higher doses.
Heparin’s anticoagulant effect is immediate when administered IV, but there is a delay of up to 60 minutes when given SC. The drug’s elimination half-life ranges from 40 to 90 minutes. In urgent clinical situations, treatment often begins with a bolus intravenous dose followed by a continuous infusion. The activated Partial Thromboplastin Time (aPTT) is often used to monitor the effectiveness of the treatment. Time to clot formation is measured in seconds using aPTT. Partial Thromboplastin Time and activated Partial Thromboplastin Time measure the same thing – the actual time it takes for the blood to clot. The reference ranges are different for aPTT and PTT. This is because an activator (aPTT) is added to speed up the clotting time and narrow the reference range, making it more sensitive than PPT.
- Reference range for aPTT is 30-40 seconds.
- Reference range for PTT is 60-70 seconds.
Therapeutic aPTT is usually 1.5 – 2.5 times the reference range. aPTT is measured and heparin dose adjusted every 6 hours. Once stable, aPTT is measured daily.
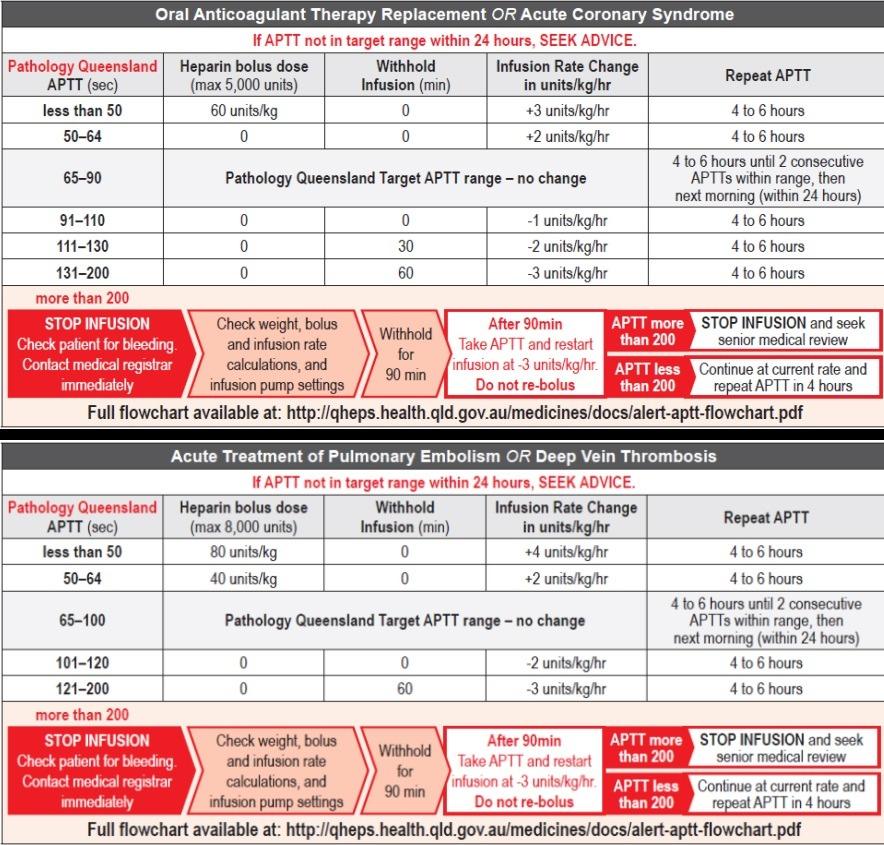
The tables below are sourced from the Qld Health Heparin Infusion Order Form. Note that the target range differs between indications. This highlights the importance of particular caution when dealing with heparin infusions.
LMWHs administered SC, have a longer and dose-independent elimination half-life, following first-order kinetics. This predictability allows for less frequent dosing, typically once or twice daily, without the need for routine monitoring through APTT, unlike UFH. LMWHs are primarily excreted by the kidneys, making UFH a preferred option in cases of renal failure. However, except for this situation, LMWHs are equally safe and effective as UFH, offering the convenience of self-administration at home without the need for frequent blood tests or dose adjustments.
Drug interactions with the heparins include any drugs that potentially affect the clotting process, and current drug information sources should be consulted. In addition, the heparins can cause hyperkalaemia, and combinations with any drugs that increase this risk should be avoided or the plasma potassium concentration should be monitored.Adverse effects with the heparins include bleeding or haemorrhage, thrombosis (uncommon but serious), osteoporosis, hyperkaelemia, hypoaldosteronism and local irritation effects such as erythema, haematomas, urticaria and pain at the injection site. The incidence of thrombocytopenia is less (around 0.6%).The clinical indications of heparin and LMWH are listed below:
- Prevention of VTE in surgical and high-risk medical patients.
- Prevention of VTE in surgical patients (sole indication for danaparoid).
- Prevention of clotting during haemodialysis.
- Treatment of DVT.
- Treatment of acute STEMI, non-STEMI and unstable angina (enoxaparin).
Watch the brief vodcast on the anticoagulant, Heparin (12 minutes)
Warfarin
Warfarin’s discovery traces back to the 1920s when agricultural shifts in North America led to unexpected outcomes. The switch from corn to sweet clover in cattle feed resulted in a widespread hemorrhagic disease among cattle. This issue was eventually linked to bishydroxycoumarin present in spoiled sweet clover, which paved the way for the development of warfarin. It is named after the Wisconsin Alumni Research Foundation. Initially, warfarin was used as a rodenticide, but it soon became a leading anticoagulant for managing and preventing thromboembolic disorders.
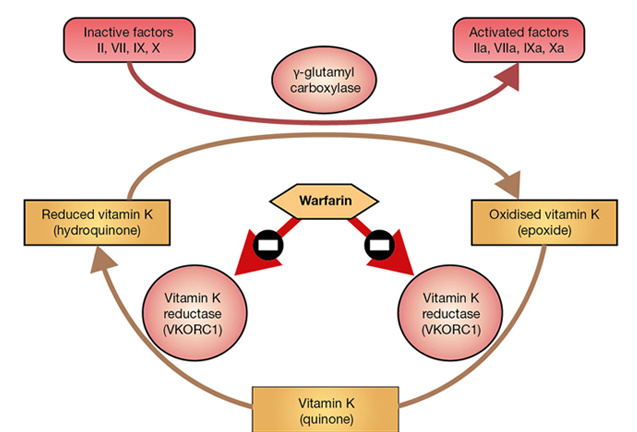
Warfarin is a vitamin K antagonist. Warfarin interferes with hepatic synthesis of the vitamin K–dependent clotting factors through inhibition of the vitamin K epoxide reductase complex 1 (VKORC1). As a consequence, interference in the γ-carboxylation of the glutamic acid residues on factors II, VII, IX and X, and on various anticoagulant proteins by γ-glutamyl carboxylase, leads to the production of non-functional coagulation factors. Warfarin has no direct action on the coagulation factors already present in the blood, so the effects of warfarin develop as the factors are cleared. Factor VII is depleted quickly; the sequential depletion of factors IX, X and II follows. The half lives of the factors are
- Factor VII and protein C – 6-8hrs
- Factor IX –24 hrs
- Factor X – 36 hrs
- Factor II – 54hrs
- Protein S – 30hrs
Therefore, UFH and LMWH therapy should be continued for at least 48 hours after warfarin is commenced and the INR reaches the therapeutic range.
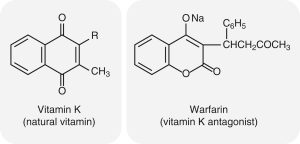
The following images shows the chemical structures of Vitamin K and warfarin. Warfarin, a vitamin K antagonist, is an oral anticoagulant. It competes with vitamin K (note the similarity in their structures) for the reductase enzyme (vitamin K epoxide reductase component 1 [VKORC1]) that activates vitamin K and is the site of its action.
 Warfarin does not affect established clots but prevents further extension of formed clots, thereby diminishing the potential for secondary thromboembolic complications. The main advantage of warfarin is that it is effective orally and can be given once daily after the maintenance dose has been established. However, despite its long-standing role in medicine, by 2018, DOACs had begun to surpass warfarin. This is because warfarin has a low margin of safety and requires frequent blood tests to individualise doses, which makes it inconvenient for patients.
Warfarin does not affect established clots but prevents further extension of formed clots, thereby diminishing the potential for secondary thromboembolic complications. The main advantage of warfarin is that it is effective orally and can be given once daily after the maintenance dose has been established. However, despite its long-standing role in medicine, by 2018, DOACs had begun to surpass warfarin. This is because warfarin has a low margin of safety and requires frequent blood tests to individualise doses, which makes it inconvenient for patients.
Warfarin is a racemic compound that includes equal amounts of S- and R-warfarin. It is efficiently absorbed from the gastrointestinal tract, achieving over 95% systemic bioavailability. The peak plasma concentration is reached within 3 to 9 hours, and the drug’s effects last from 2 to 5 days. Warfarin is extensively bound to plasma proteins (99%), and its plasma half-life ranges from 25 to 60 hours, with an average of 40 hours. The drug can cross the placenta, leading to fetal plasma concentrations similar to those in the maternal blood.
S-warfarin is primarily metabolized in the liver by CYP2C9, with minor contributions from CYP2C8, CYP2C18, and CYP2C19. The significant role of CYP2C9 in the metabolism of S-warfarin explains the considerable variability in warfarin dosing requirements. Individuals with certain CYP2C9 variant alleles may experience reduced metabolism of S-warfarin, resulting in higher plasma concentrations of the drug. This genetic variation is notably present in European, African, and Asian populations. There is limited evidence suggesting that genetic testing improves anticoagulation management or reduces bleeding risks. In comparison, R-warfarin is metabolized mainly by CYP1A2 and CYP3A4, with minor involvement from CYP1A1, CYP2C8, CYP2C18, CYP2C19, and CYP3A5.
Numerous drugs, including antimicrobials, cardiovascular drugs, analgesics, anti-inflammatory drugs, immunomodulators, gastrointestinal drugs, antineoplastics and central nervous system drugs and St John’s wort interact with warfarin, and current drug information resources should always be consulted.
Examples of drugs decreasing the anticoagulant effect include:
- St John’s Wort, carbamazepine, barbiturates, rifampicin, phenytoin (can also rarely increase anticoagulant effect)
Examples of drugs increasing the anticoagulant effect include:
- Antifungals and various antibiotics, thyroid hormones, statins, aspirin, NSAIDS, paracetamol and SSRIs.
N.B. Paracetamol – widely used as an analgesic for patients taking warfarin and is preferred to aspirin and COX-2 inhibitors (increased risk of bleeding). Small occasional doses of paracetamol do not usually alter anticoagulant effect of warfarin. Larger doses of paracetamol (2-4g daily for several days (therapeutic guidelines) or 3.5-7g per week (AMH 2008)) may Increase INR.
If drug-drug interactions exist, consider substituting an interacting drug with a non-interacting drug if possible. Alternatively, monitor INR and increase/decrease warfarin dose accordingly (interacting drug should be taken regularly as PRN use would not allow INR to stabilise).
Warfarin is usually commencement doses of 5 mg once daily for 2 days and then the dose is adjusted according to INR. INR is an international normalized ratio that provides standardized reporting of the actual prothrombin time (PT). This allows comparisons to be made between different labs, hospitals and countries. Warfarin use represents a balance between thrombosis (lower end of the therapeutic range) and haemorrhage (upper end of the therapeutic range). There is a strong relationship between INR and bleeding risk (especially if INR is over 5 – 6). A typical dose of warfarin can sit anywhere between 1 and 10mg once daily – this is needed to achieve the recommended INR ranges as follows:
- 2.0 – 3.0 AF, DVT, PE and bio-prosthetic heart valves in patients with SR for 6 weeks post-op
- 2.5 – 3.5 Mechanical prosthetic heart valves
The main adverse effects of warfarin include bleeding or haemorrhage (very common), and rarely skin necrosis, calciphylaxis, purple discolouration of toes, alopecia, fever, rash, nausea, vomiting, diarrhoea, hepatic dysfunction and allergic reactions. If the patient is over-anticoagulated, the antidote Vitamin K can be administered.
Precautions for the use of warfarin are listed below:
- Avoid use in the elderly. Risk factors for bleeding include age older than 70 years, previous history of stroke and falls, liver disease, chronic renal failure, drug interactions and evidence of gastrointestinal bleeding in the previous 18 months.
- Contraindicated in alcoholics.
- Avoid use in pregnancy (except for women with prosthetic heart valves). Risk of abortion and teratogenicity is high, and fetal abnormalities and facial anomalies have been reported if warfarin is administered during the first trimester. Administration during the second and third trimesters is associated with central nervous system abnormalities.
- Avoid use in people with known anticoagulant drug hypersensitivity, any medical or surgical condition associated with bleeding (aneurysm, cerebrovascular bleeding, surgery and severe trauma), blood disorders, severe uncontrolled hypertension, pericarditis, severe diabetes, ulcers, visceral cancer, vitamin C or vitamin K deficiencies, endocarditis or severe liver or kidney impairment.
- Brands of warfarin should not be interchanged due to a lack of bioequivalence data.
Important counselling points about warfarin include:
- Take the tablets at about the same time each day. Use a calendar or ‘anticoagulant book’ to keep a record of your dose, and mark off the date immediately after taking a dose.
- If you forget to take a dose, notify the doctor immediately. Take the dose as soon as possible on the same day. Do not take a double dose the next day to make up for a missed dose.
- If you develop signs of bleeding (e.g. bleeding gums, prolonged bleeding from cuts, black and tarry stools, nosebleeds, unusual bruising); purple colouration, pain or tenderness in the toes; sudden, intense pain in a leg or foot; a painful, bruise-like rash; headaches; dizziness; or weakness, seek immediate medical attention.
- There are two brands of warfarin; do not swap from one brand to another.
- You will need to have regular blood tests while you are taking this medicine to measure your rate of blood clotting; your doctor will then tell you what dose to take. Tell your doctor if you have any other illness (e.g. diarrhoea, vomiting, infection, fever) because you may require extra blood tests.
- Tell your doctor, pharmacist, dentist and other health professionals that you are taking warfarin.
- Do not take any other prescription or over-the-counter medicines or herbal products without medical advice.
- Maintain a normal, balanced diet and limit alcohol intake. Do not make major changes to your diet because certain foods contain vitamin K, which can interfere with the effect of warfarin.
- Consider wearing a Medic Alert bracelet and carrying a card showing the details of your treatment.
The clinical indications for warfarin are listed below:
- Prevention of VTE
- Treatment of VTE
- Prevention of thromboembolism in patients with prosthetic heart valves
- Prevention of stroke in patients with previous MI and increased embolic risk
- AF and a high risk of stroke or systemic embolism.
Direct Thrombin Inhibitors
The use of medicinal leeches (Hirudo medicinalis) dates back over 2,500 years. In 1884, it was found that blood within the leech’s gut remained uncoagulated, leading to the isolation of the anticoagulant substance hirudin from the leech’s pharyngeal glands in 1957 by Markwardt. Hirudin functions as a direct, irreversible, non-covalent inhibitor of thrombin, though its anticoagulant effects can be variable.
Bivalirudin is a synthetic polypeptide consisting of 20 amino acids, designed as a hirudin analogue. It binds directly and reversibly to thrombin, bypassing the need for antithrombin III (AT III) and thereby inhibiting thrombin’s clot-forming actions. Due to its rapid dissociation from thrombin, a portion of thrombin remains active, which is important for maintaining hemostasis. With a plasma half-life of about 25 minutes, bivalirudin is typically administered via intravenous bolus and continuous infusion. Its use requires careful consideration of bleeding risks and renal function, as severe bleeding is a common side effect, and anaphylaxis has been occasionally reported. It is indicated in moderate-to-high risk unstable angina and non-ST-segment elevation MI (NSTEACS) undergoing early invasive management, including PCI.
Dabigatran etexilate is an oral medication that directly inhibits thrombin. Due to dabigatran’s low oral bioavailability (~7%), dabigatran etexilate, the prodrug form, enhances gastrointestinal absorption. After conversion to dabigatran by hepatic carboxylesterases, this active form specifically and competitively inhibits both free and clot-bound thrombin by interacting with its active site. The antidote for dabigatran is idarucizumab, a humanised monoclonal antibody fragment that binds dabigatran and its metabolites, blocking its effect.
Dabigatran etexilate is indicated for:
- Prevention of VTE after elective total hip or knee replacement
- Treatment of acute VTE and prevention of subsequent VTE
- Non-valvular AF and a high risk of stroke or systemic embolism.
Factor Xa Inhibitors
The two most commonly prescribed factor Xa inhibitors are rivaroxaban and apixaban which are administered orally. Fondaparinux is administered via the SC route. 
Rivaroxaban, following dabigatran etexilate, was the second novel oral anticoagulant (NOAC) introduced after warfarin. It acts as a direct, dose-dependent reversible inhibitor by competitively binding to the active site of factor Xa, which is involved in both the intrinsic and extrinsic coagulation pathways. This action reduces thrombin production and prevents the conversion of fibrinogen to fibrin, thereby inhibiting clot formation. Rivaroxaban leads to the prolongation of prothrombin time (PT) and activated partial thromboplastin time (aPTT), with PT prolongation being directly related to the plasma concentration of the drug. The absorption of rivaroxaban is rapid, with bioavailability ranging between 60% and 80%. Around 30% of the administered dose is excreted unchanged through urine, while the remaining portion is metabolized via the CYP3A4, CYP2C8, and other CYP-independent pathways.
Apixaban, the most recent addition to the NOACs, also directly inhibits factor Xa, similar to rivaroxaban, and is primarily used for the treatment of venous thromboembolism (VTE) following elective knee or hip replacement surgery. Apixaban is quickly absorbed with a bioavailability of approximately 50%. Like rivaroxaban, it is a substrate for P-glycoprotein (P-gp) and is metabolized mainly through demethylation and hydroxylation by CYP3A4/5, with lesser contributions from CYP1A2, 2C8, 2C9, 2C19, and CYP2J2. The elimination of apixaban occurs via multiple routes: around 25% is excreted unchanged in the urine, another 25% as metabolites primarily through feces, and some evidence suggests direct biliary excretion. Similar to rivaroxaban, the use of strong CYP3A4 and P-gp inhibitors is contraindicated, as they may significantly affect the drug’s plasma levels. Conversely, CYP3A4 and P-gp inducers, such as carbamazepine, rifampicin, phenytoin, phenobarbital, and St. John’s wort, can lower apixaban concentrations, reducing its clinical effectiveness. Routine monitoring of anticoagulant effects is not required, thus there is no standard method for dosage adjustment. However, in cases of severe renal impairment (CrCl 15–29 mL/min), an increase in plasma drug concentration has been observed, necessitating dosage adjustments.
Both rivaroxaban and apixaban carry a risk of bleeding, a common adverse effect. Andexanet alfa, a modified human factor Xa, serves as the antidote for these anticoagulants.
Watch the brief vodcast on other anticoagulants, warfarin, direct thrombin inhibitors and factor Xa inhibitors (16 minutes)
You will remember the process of platelet activation from the previous chapter. Upon activation, platelets undergo a series of essential reactions that contribute to haemostasis, assist in repairing damaged blood vessels, and participate in inflammation. Many of these reactions are interconnected, with multiple pathways available to compensate if one is inhibited, and several are self-amplifying, providing positive feedback. If you don’t remember the process of platelet process, we will quickly recap!
- Adhesion after vascular injury: von Willebrand factor acts as a bridge between subendothelial macromolecules and GPIb receptors on the platelet surface.
- Shape transformation: Platelets change from smooth discs to spiny spheres with pseudopodia.
- Granule secretion: Platelets release contents like ADP, 5-hydroxytryptamine, coagulation factors, and growth factors, including platelet-derived growth factor (PDGF).
- Synthesis of labile mediators: Platelets produce mediators such as platelet-activating factor and thromboxane A2.
- Aggregation: Various agonists, including collagen, thrombin, ADP, 5-hydroxytryptamine, and TXA2, promote platelet aggregation by interacting with specific receptors, leading to the expression of GPIIb/IIIa receptors that bind fibrinogen, connecting adjacent platelets into aggregates.
- Exposure of acidic phospholipids (PL): This promotes thrombin formation and further platelet activation through thrombin receptors, as well as fibrin formation by cleaving fibrinogen.
These processes are vital for haemostasis but can be inadvertently triggered in diseased artery walls, particularly in the presence of atherosclerosis, leading to thrombosis.
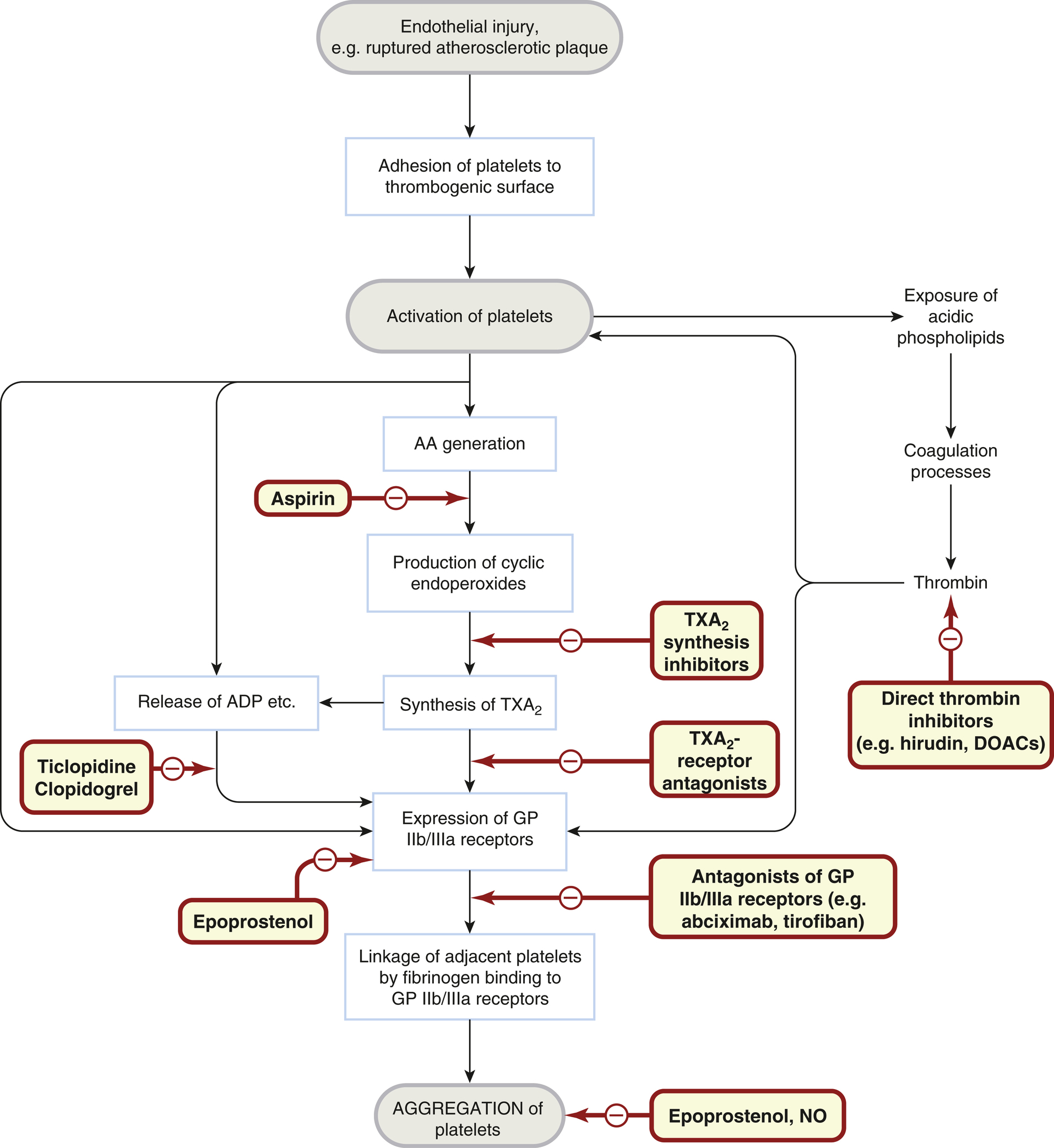
Platelets play such a critical role in thromboembolic disease that it is no surprise that antiplatelet drugs are of great therapeutic value. Clinical trials of aspirin radically altered clinical practice, and more recently drugs that block ADP receptors and GPIIb/IIIa have also been found to be therapeutically useful. Antiplatelet drugs are used to treat arterial thrombosis and include:
- Aspirin and dipyridamole
- P2Y12 inhibitors clopidogrel, prasugrel and ticagrelor
- Glycoprotein IIb/IIIa receptor inhibitors eptifibatide and tirofiban.
Aspirin
Aspirin exerts a prolonged inhibitory effect on platelet function by permanently blocking the cyclooxygenase enzyme COX-1, which is essential for the production of thromboxane A2. Thromboxane A2 plays a critical role in promoting platelet aggregation and vasoconstriction, so aspirin effectively reduces these processes. Since platelets cannot regenerate COX-1, the impact of aspirin persists for 8–10 days, the time it takes for new platelets to form. This mechanism underlies aspirin’s utility as an antiplatelet agent and its tendency to extend bleeding time.
Extensive research has demonstrated that aspirin therapy significantly lowers mortality in individuals experiencing acute myocardial infarction. It is a cornerstone treatment for both the primary and secondary prevention of cardiovascular and cerebrovascular diseases. The antiplatelet effects of aspirin are achieved with daily doses ranging from 75 to 300 mg, with no added benefits observed at higher doses. Low-dose aspirin (75–150 mg/day) does not alter bleeding time. Aspirin is also available in combination with clopidogrel (clopidogrel 75 mg/aspirin 100 mg) or dipyridamole (dipyridamole 200 mg [controlled release]/aspirin 25 mg) in fixed-dose formulations.
In some individuals taking long-term low-dose aspirin, the risk of thrombotic events persists due to incomplete inhibition of platelet aggregation, a phenomenon known as ‘aspirin resistance.’ To address this issue, strategies include avoiding drugs that counteract aspirin’s effects, such as non-steroidal anti-inflammatory drugs (NSAIDs), and considering a twice-daily dosing regime.
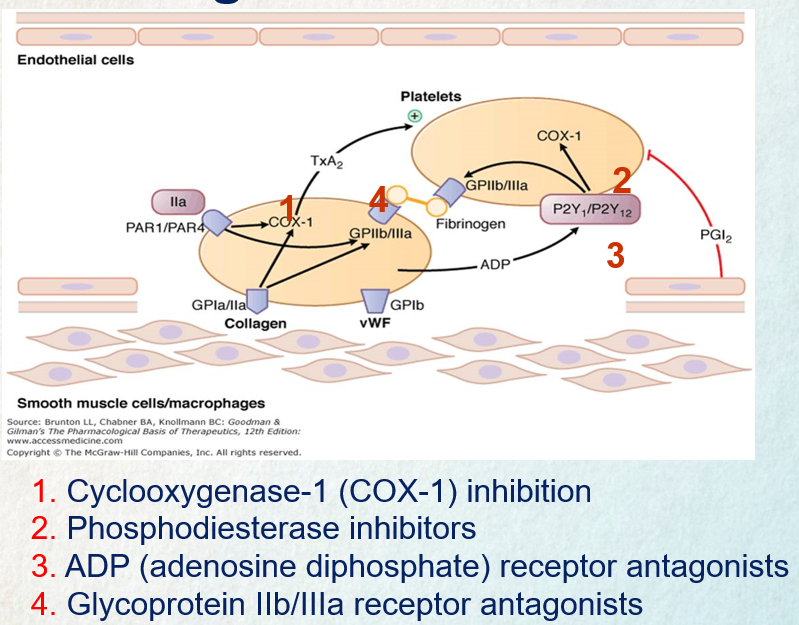
P2Y12 inhibitors
P2Y12 inhibitors work by blocking ADP-induced platelet aggregation. This is achieved by binding to the P2Y12 receptor on platelets, either irreversibly (as seen with clopidogrel and prasugrel) or reversibly (as with ticagrelor). This binding action prevents the activation of the glycoprotein IIb/IIIa complex by ADP, thereby inhibiting platelet aggregation. Once P2Y12 receptors are blocked, they lose the ability to interact with ADP for the lifespan of the platelet. Upon stopping the medication, normal platelet function typically resumes within approximately one week.
Clopidogrel, a second-generation thienopyridine, is a prodrug that undergoes extensive metabolism. Around 85% is converted to an inactive carboxylic acid metabolite, while the remaining 15% is metabolized into an active form through a two-step process involving several cytochrome P450 enzymes, including CYP2C19, CYP1A2, CYP2B6, and CYP3A. The active metabolite reaches its peak concentration roughly one hour after administration. Unlike aspirin, clopidogrel’s active metabolite does not affect prostaglandin synthesis. Response variability to clopidogrel has been observed, likely due to genetic factors like CYP2C19 polymorphisms and clinical factors such as medication adherence. This variability has led to the development of more potent, third-generation P2Y12 inhibitors like prasugrel and ticagrelor.
Prasugrel is also a prodrug that is quickly absorbed and activated. It undergoes hydrolysis in the intestine to form an intermediate compound (R-95913), which is further metabolized in the liver to the active metabolite R-138727 via CYP3A4, CYP2B6, CYP2C9, and CYP2C19. This active metabolite is then broken down into inactive forms that are excreted in the urine. The drug reaches peak concentration within 30 minutes, with a rapid onset of action that is more potent and consistent than clopidogrel. Although the active metabolite’s half-life is around 4 hours, prasugrel’s irreversible binding to the P2Y12 receptor results in prolonged action even after stopping the drug.
Ticagrelor, in contrast, is not a prodrug and binds reversibly to the P2Y12 receptor. It is absorbed quickly after oral intake, reaching peak plasma levels in 1.5 to 3 hours. Ticagrelor is metabolized by CYP3A4 to form an active metabolite (AR-C124910XX), which accounts for about 40% of the drug’s plasma concentration. Ticagrelor provides more consistent and potent platelet aggregation inhibition compared to clopidogrel. It is important to avoid co-administration with strong CYP3A4 inhibitors or inducers, and to monitor digoxin levels since ticagrelor inhibits P-glycoprotein.
Bleeding is a common and potentially serious adverse effect associated with P2Y12 inhibitors. These medications are contraindicated in patients with active bleeding, a history of stroke or transient ischemic attack, bleeding disorders, or severe liver disease. Prasugrel is generally not recommended for individuals over 75 years old. Caution is advised when using ticagrelor in patients with cardiac arrhythmias, especially bradycardia, advanced atrioventricular block, or sick sinus syndrome. Additionally, ticagrelor may exacerbate asthma or chronic obstructive pulmonary disease (COPD) and can worsen hyperuricemia.
Clinical Uses of P2Y12 Inhibitors incloude:
- Acute coronary syndrome (in combination with aspirin)
- Stent implantation (prasugrel with aspirin)
- Prevention of thromboembolism in patients with ACS (clopidogrel).
Glycoprotein IIb/IIIa receptor inhibitors
The inhibition of glycoprotein IIb/IIIa receptors disrupts the final step of platelet aggregation by preventing fibrinogen from binding to these receptors. This process is crucial in the formation of platelet clots, and blocking it effectively reduces the potential for clot formation.
Eptifibatide and tirofiban, both of which are peptides, are designed to mimic a specific sequence found in glycoprotein IIb/IIIa receptors. These drugs are administered parenterally and are typically used in conjunction with low-dose aspirin or heparin to minimize the risk of ischemic complications during procedures like percutaneous transluminal coronary angioplasty or intracoronary stenting. Attempts to create effective oral versions of these drugs have not yet been successful.
Drug interactions with glycoprotein IIb/IIIa receptor inhibitors inlucde:
- Oral Anticoagulants: Use of glycoprotein IIb/IIIa inhibitors within a week of oral anticoagulants is discouraged due to a heightened risk of bleeding.
- Antiplatelet Drugs (e.g., Dipyridamole): If used together, there is an increased risk of bleeding, necessitating careful monitoring.
- Dextran, Low Molecular Weight Heparins (LMWHs), NSAIDs: Concurrent use increases the likelihood of bleeding and hemorrhage.
- Thrombolytic Drugs: Combining or sequentially administering these drugs with glycoprotein IIb/IIIa inhibitors can also increase bleeding risk, so close monitoring is recommended.
Common adverse effects include both minor and major bleeding, thrombocytopenia, visual disturbances, confusion, nausea, vomiting, and hypotension. It is essential to monitor parameters such as PT (Prothrombin Time), aPTT, creatinine clearance, platelet count, hemoglobin, and hematocrit levels before and during treatment.
You should known that the main antiplatelet drug is aspirin. Other drugs with distinct actions (e.g. dipyridamole, clopidogrel, ticagrelor) can have additive effects, or be used in patients who are intolerant of aspirin. Uses of antiplatelet drugs relate mainly to arterial thrombosis and include:
- Acute myocardial infarction
- Prevention of myocardial infarction in patients at high risk, including a history of myocardial infarction, angina or intermittent claudication
- Following coronary artery bypass grafting
- Unstable coronary syndromes (a P2Y12 antagonist such as clopidogrel, prasugrel or ticagrelor is added to aspirin)
- Following coronary artery angioplasty and/or stenting. Intravenous glycoprotein IIb/IIIa antagonists, e.g. abciximab, are used in some patients in addition to aspirin.
- Transient cerebral ischaemic attack (‘ministrokes’) or thrombotic stroke, to prevent recurrence (dipyridamole can be added to aspirin)
- Atrial fibrillation, if oral anticoagulation is contraindicated; or, by specialists, in high-risk situations in combination with anticoagulant.
Watch the brief vodcast on antiplatelet drugs (12 minutes)
Thrombolytics/Fibrinolytics
When the coagulation system is triggered, the fibrinolytic system is simultaneously activated through various endogenous plasminogen activators such as tissue plasminogen activator (tPA), urokinase-type plasminogen activator, kallikrein, and neutrophil elastase. tPA is inhibited by lipoprotein(a), a structurally similar lipoprotein, elevated levels of which are considered an independent risk factor for myocardial infarction. Plasminogen is deposited on the fibrin strands within a thrombus. Plasminogen activators are serine proteases and are unstable in circulating blood. They infiltrate the thrombus and convert plasminogen, a plasma zymogen, into plasmin at the site. Plasmin, a protease resembling trypsin, breaks down fibrin as well as fibrinogen, factors II, V, and VIII, and various other proteins. Any plasmin that enters the circulation is neutralized by plasmin inhibitors, such as PAI-1, which prevent internal self-digestion.
Drugs can influence this system by either promoting or inhibiting fibrinolysis, leading to fibrinolytic or antifibrinolytic effects, respectively.
The following figure illustrates the interaction of the fibrinolytic system with the coagulation cascade and platelet activation, and the action of drugs that modify this. Several fibrinolytic (thrombolytic) drugs are used clinically, principally to reopen the occluded arteries in patients with acute myocardial infarction or stroke, less commonly in patients with life-threatening venous thrombosis or pulmonary embolism.
Streptokinase, a protein that activates plasminogen, is derived from Streptococcal cultures. When administered intravenously, it can lower mortality in cases of acute myocardial infarction, with its positive effects being enhanced when combined with aspirin. However, the action of streptokinase can be inhibited by antibodies that develop four days or more after the initial dose, meaning that its use should not be repeated beyond this time frame.
Alteplase and duteplase are recombinant forms of tissue plasminogen activator (tPA), with the former being a single-chain version and the latter a double-chain version. These drugs are more effective on plasminogen bound to fibrin rather than in plasma, making them “clot selective.” Unlike streptokinase, recombinant tPA does not trigger an immune response and can be used in patients with antibodies to streptokinase. Due to their brief half-lives, both alteplase and duteplase require intravenous infusions. Reteplase, which is similar to these drugs but has a longer half-life, can be administered in bolus form, simplifying its use in treating myocardial infarction.
Fibrinolytic agents pose a significant risk of bleeding, which can manifest as gastrointestinal hemorrhage or hemorrhagic stroke. In severe cases, bleeding may be managed with treatments like tranexamic acid (antifibrinolytic), fresh plasma, or coagulation factors. Tranexamic acid inhibits plasminogen activation and thus prevents fibrinolysis. Streptokinase, in particular, is known to cause allergic reactions and mild fever. It also leads to a surge in plasmin production, which releases vasodilator kinins and may result in hypotension.
These agents should not be used in cases of active internal bleeding, hemorrhagic cerebrovascular disease, bleeding disorders, pregnancy, uncontrolled hypertension, procedures requiring strict hemostasis, or recent trauma, including rigorous cardiopulmonary resuscitation.
You should know that the main drugs are tPAs, for example, alteplase. Clinical indications of thrombolytics/fibrinolyitcs include:
- The main use is in acute myocardial infarction, within 12 h of onset (the earlier the better!).
- Other uses include:
- acute thrombotic stroke within 3 h of onset (tPA), in selected patients
- clearing thrombosed shunts and cannulae
- acute arterial thromboembolism
- life-threatening deep vein thrombosis and pulmonary embolism (streptokinase, given
Watch the brief vodcast on the thrombolytics and conclusion (7 minutes)
In summary
- Classes of Antithrombotic Drugs:
- Anticoagulants: Inhibit fibrin formation, prevent clot growth, and reduce thromboembolic event risks.
- Antiplatelet Drugs: Prevent platelet aggregation, reducing the risk of arterial thrombi.
- Thrombolytics/Fibrinolytics: Dissolve existing clots by breaking down fibrin.
- Heparin indirectly blocks the action of thrombin and Factor Xa via the increased activation of Antithrombin III. In contrast, the LMWHs block Factor Xa by activating Antithrombin III but have little effect at thrombin.
- The direct thrombin inhibitors directly bind to thrombin and block it’s effect. Because thrombin has both anticoagulant actions and also activates platelets – the DTIs have both antiplatelet and anticoagulant effects. They have a rapid onset and specific antidotes.
- The Factor Xa inhibitors selectively inhibit Factor Xa blocking thrombin production. This reduces fibrin and thrombus formation. There are antidotes available for bleeding management.
- Warfarin, the only Vitamin K antagonist blocks the effect of Vitamin K reductase. This reduces the activation of the vitamin K dependent clotting factors II, VII, IX and X. No effect on factors already present in the blood. Onset of action depends on clearance of those factors already present. Dose monitoring and low safety profile means largely replaced by DOACs.
- The antiplatelet drugs have various targets on the platelet surface including P2Y12, Glycoprotein IIb/IIIa, COX-1 and others.
- The thrombolytics are not anticoagulants. Rather they break down formed clots by converting plasminogen to plasmin which then catalyses the breakdown of fibrin. hese drugs promote fibrinolysis to dissolve clots in acute myocardial infarction and stroke but carry significant bleeding risks and are contraindicated in several conditions.
- All drugs which affect coagulation have potential adverse effects, particularly bleeding, and should be used with caution in patients with contraindications such as active bleeding or recent trauma.